Peter C. Gøtzschea and David Healyb
International Journal of Risk & Safety in Medicine – 2022
Traduzione in italiano a cura del Dott. Claudio Ajmone per GiùleManidaiBambini.org
Testo originale in inglese, disponibile a questo link
aInstitute for Scientific Freedom, Hørsholm, Denmark
bDepartment of Family Medicine, McMaster University, Hamilton, Canada
Abstract
BACKGROUND: La fluoxetina è stata approvata per la depressione nei bambini e negli adolescenti sulla base di due studi controllati con placebo, X065 e HCJE, con 96 e 219 partecipanti, rispettivamente.
OBIETTIVO: rivedere questi studi, che sembrano essere stati riportati in modo errato.
METODI: revisione sistematica dei rapporti e delle pubblicazioni degli studi clinici. Gli esiti primari erano le variabili di efficacia previste dai protocolli di studio, gli eventi suicidari e i precursori della suicidalità o della violenza.
RISULTATI: mancavano informazioni essenziali e vi erano incongruenze numeriche inspiegabili. (1) I risultati di efficacia sono stati distorti a favore della fluoxetina a causa dei differenti abbandoni e dei dati mancanti. L’efficacia della Children’s Depression Rating Scale-Revised era pari al 4% del punteggio basale, un valore non clinicamente rilevante. Le valutazioni dei pazienti non hanno evidenziato l’effcacia della fluoxetina. (2) Nelle pubblicazioni e nei rapporti di studio mancavano eventi suicidari. I precursori della suicidalità o della violenza si sono verificati più spesso con la fluoxetina che con il placebo. Per lo studio HCJE, il numero necessario di danni era di 6 per gli eventi del sistema nervoso, 7 per i danni moderati o gravi e 10 per i danni gravi. La fluoxetina ha ridotto l’altezza e il peso per 19 settimane rispettivamente di 1,0 cm e 1,1 kg e ha prolungato l’intervallo QT.
CONCLUSIONI: La nostra rianalisi dei due studi cardine ha dimostrato che la fluoxetina non è sicura e non è efficace.
1. Introduzione
La fluoxetina è stata approvata per la depressione nei bambini e negli adolescenti negli Stati Uniti nel 2002, sulla base di due studi clinici controllati con placebo, anche se una revisione statistica per la Food and Drug Administration (FDA) aveva rilevato che non vi era un beneficio statisticamente significativo del farmaco sull’esito primario in nessuno dei due studi [1].
Nel 2004, l’FDA ha emesso un’avvertenza “black box” secondo cui tutti gli antidepressivi possono aumentare il rischio di pensieri e comportamenti suicidi nei bambini e negli adolescenti. Un aumento degli eventi suicidari è stato osservato anche negli adulti. Una meta-analisi di studi controllati con placebo condotti su volontari adulti sani, utilizzando i precursori di eventi definiti dall’FDA, ha rilevato che gli SSRI e gli SNRI raddoppiano il rischio di danni legati alla suicidalità e alla violenza, mentre il numero necessario da trattare per danneggiare una persona sana è solo 16 (intervallo di confidenza al 95% da 8 a 100) [2].
Circa la metà dei suicidi non compare negli studi pubblicati sugli psicofarmaci [3], e gli eventi suicidari sono spesso chiamati in un altro modo, ad esempio labilità emotiva, ricovero in ospedale o depressione [4-6]. Nessuna delle pubblicazioni peer-reviewed [7,8] ha descritto gli eventi suicidari menzionati nelle relazioni sugli studi clinici (CSR), X065 e HCJE, che Eli Lilly ha presentato agli enti regolatori del farmaco per l’approvazione della commercializzazione [9,10]. Inoltre, Lilly ha concluso che la fluoxetina era efficace, anche se l’FDA ha riscontrato che entrambi gli studi erano negativi per quanto riguarda l’endpoint primario. Abbiamo quindi deciso di rivedere e ripristinare la documentazione pubblica degli studi [4-6].
2. Metodi
Abbiamo ottenuto le CSR dei due studi dalla UK Medicines and Healthcare products Regulatory Agency. Come previsto dalla metodologia RIAT, abbiamo chiesto a Eli Lilly e a Graham Emslie, lo sperimentatore principale, se volevano ripristinare gli studi [11]. Emslie non ci ha risposto. Lilly non ritiene che “in questo momento siano necessarie ulteriori analisi”.
Le CSR per X065 e HCJE sono rispettivamente di 1008 e 2549 pagine [9,10]. Ci sono state riduzioni, soprattutto di nomi di persone, finanziatori e istituzioni. Molte sezioni erano vuote anche se gli indici suggerivano il contrario. Le sezioni vuote avrebbero dovuto includere l’anamnesi psichiatrica, i dati di efficacia, gli eventi avversi e i dati dell’elettrocardiogramma, ma consistevano in una pagina con il testo: “Si prega di consultare il modulo di trasporto SAS che si trova al punto 11 della presente documentazione”, che non era presente.
Il referto X065 conteneva 159 pagine con i dati dei singoli pazienti per i valori di laboratorio anormali. Il rapporto HCJE conteneva 535 pagine con dati di laboratorio e dati dei singoli pazienti relativi alle concentrazioni ematiche di fluoxetina e norfluoxetina, con dettagli su peso, età, altezza, indice di massa corporea, origine etnica e assunzione di fumo, alcol e caffeina.
Mancavano molte pagine. L’indice di X065 conteneva voci per le pagine 833 e 869, ma non segnalava che le pagine da 841 a 868 non esistevano. In HCJE, un indice secondario a pagina 1471 non indicava che mancasse qualcosa perché non c’erano numeri di pagina, ma solo titoli di sezione, ma mancavano 470 pagine. I materiali mancanti rendono impossibile un restauro completo del RIAT.
Gli esiti primari erano le variabili di efficacia elencate come primarie nei protocolli di studio, nonché gli eventi suicidari e i precursori del suicidio o della violenza. Abbiamo confrontato gli esiti riferiti dai pazienti con quelli riferiti dagli sperimentatori. Ci siamo concentrati sull’accecamento, sugli effetti dell’astinenza nei pazienti che avevano interrotto un farmaco antidepressivo prima della randomizzazione e sull’uso concomitante di farmaci con proprietà sedative che potrebbero oscurare danni come l’agitazione con la fluoxetina [4,5].
Uno sperimentatore (PCG) ha estratto i termini per tutti i dati sugli eventi avversi, li ha ordinati alfabeticamente e li ha presentati all’altro sperimentatore (DH) che ha deciso in cieco, senza sapere se si fossero verificati con la fluoxetina o con il placebo o quanti fossero, se potessero essere considerati precursori di eventi suicidi o violenti. Le opzioni di risposta erano no, forse e sì, con un campo di commento. Abbiamo utilizzato il test esatto di Fisher per le proporzioni. Non abbiamo eseguito meta-analisi perché i dati di efficacia erano parziali.
3. Risultati
3.1. Risultati dello studio X065
Si tratta di uno studio monocentrico avviato dallo sperimentatore e condotto in Texas dal 10 aprile 1991 al 28 febbraio 1995, pubblicato nel 1997 [7]. Il rapporto dello studio del 2000 contiene il protocollo originale dello sperimentatore e un protocollo rivisto da Lilly [9]. Il piano di analisi statistica di Lilly è stato redatto a posteriori. Lo studio comprendeva 96 pazienti ambulatoriali (44 femmine) di età compresa tra 7 e 18 anni (media 12,8) con disturbo depressivo maggiore. La durata media dell’episodio in corso era di 14 settimane. I pazienti dovevano avere un punteggio superiore a 40 nella Children’s Depression Rating Scale-Revised (CDRS-R), una scala a 17 voci con un range di punteggio da 17 a 113. I pazienti sono stati trattati per 8 settimane con fluoxetina 20 mg una volta al giorno o placebo.
3.1.1. Randomizzazione e cecità
La randomizzazione è stata stratificata per sesso ed età [9]. Non sono state fornite informazioni sulla dimensione dei blocchi. L’assegnazione del trattamento è stata effettuata da un farmacista locale utilizzando una lista preparata dal biostatistico. Un’infermiera del sito di studio ha verificato la correttezza dell’assegnazione confrontando i farmaci dispensati con la lista. Ciò suggerisce che l’assegnazione non era nascosta. Inizialmente, la farmacia ha assegnato i farmaci in cieco svuotando le capsule di Prozac per il gruppo placebo e riempiendole nuovamente con polvere di lattosio [9]. Dall’agosto 1993, Lilly ha fornito i farmaci in cieco in capsule bianche.
La farmacia dell’ospedale informava i laboratori di chimica dell’assegnazione del trattamento “per evitare di eseguire livelli ematici non necessari [9]”. Un’infermiera del sito di studio che ha controllato i farmaci dello studio ha fatto da collegamento tra il sito clinico e la farmacia e ha fornito valutazioni psichiatriche per due pazienti. Due pazienti che hanno tentato il suicidio con la fluoxetina “potrebbero aver visto rivelata la loro assegnazione al trattamento” e ci sono stati “alcuni casi … (meno di dieci)” in cui i registri dello studio indicavano che i medici erano stati sbloccati prima del completamento dello studio. Per i pazienti che hanno abbandonato o sono diventati depressi, il medico curante ha avuto accesso all’assegnazione del trattamento [9].
3.1.2. Esiti
Gli esiti primari valutati dagli psichiatri erano due: il miglioramento sulla scala Clinical Global Impressions (CGI) e sulla CDRS-R [7]. Questi sono stati ridotti nell’articolo pubblicato a risultati binari, molto o molto migliorati sulla scala CGI, e un CDRS-R ≤ 28. La RSI prevedeva un solo esito primario, una riduzione di almeno il 30% rispetto al basale della CDRS-R, ma tre criteri di successo: remissione (CDRS-R ≤ 28), risposta (punteggio CGI-Miglioramento di 1 o 2) e recupero (entrambi i criteri) [9]. Gli esiti secondari riportati nella RSI o nell’articolo pubblicato comprendevano:
(1) Analisi di sopravvivenza della remissione sulla scala CGI [7].
(2) analisi della varianza (ANOVA) sui punteggi settimanali della CDRS-R con l’ultima osservazione riportata (LOCF) [7]
(3) Analisi dei casi osservati [9]
(4) Variazione dei punteggi CDRS-R mediante regressione lineare sui dati disponibili [7].
(5) Analisi della covarianza utilizzando i dati disponibili [7,9]
(6) CGI-Miglioramento [9]
(7) CGI-Severità [9]
(8) Brief Psychiatric Rating Scale for Children (BPRS-C) [9]
(9) Children’s Depression Inventory (CDI) per i soggetti di età inferiore ai 13 anni e Beck Depression Inventory (BDI) per quelli di età pari o superiore ai 13 anni [9].
(10) Weinberg Screening Affective Scale (WSAS) [7]
(11) Indice di depressione di Bellevue (BID), versione per genitori e pazienti [9].
(12) Scala di valutazione globale dei bambini (CGAS) [9]
(13) Scala di valutazione globale della famiglia (FGAS) [9].
Le analisi di sottogruppo hanno incluso:
(1) analisi per sesso, età e origine dei farmaci in studio [9].
(2) riduzione del 50% della CDRS-R [9]
(3) Riduzioni del 30% e del 50% per i pazienti che completano 4 settimane di trattamento [9].
(4) Analisi di tutti e 21 gli item della BPRS-C [9]
(5) Analisi di tutti e 17 gli item del CDRS-R [9]
(6) Analisi delle somme di 3-6 item del CDRS-R denominati Subtotale dell’umore, Subtotale somatico, Subtotale soggettivo e Subtotale del comportamento (non elencati nel protocollo) [9].
(7) Un’ANOVA a misure ripetute in cui le variabili dipendenti erano i punteggi CDRS-R al basale e al post-basale [9].
Gli eventi avversi sono stati raccolti chiedendo ai pazienti se presentavano uno dei 32 sintomi della lista di controllo degli effetti collaterali (descritti come 30, ma erano 32), o uno dei 30 sintomi della lista di controllo degli effetti collaterali della fluoxetina (a partire dal gennaio 1993), e raccogliendo gli eventi avversi non richiesti [9]. Gli eventi avversi sono stati codificati secondo il metodo COSTART (Coding Symbols for a Thesaurus of Adverse Reaction Terms) da personale Lilly in cieco. Sono state effettuate analisi di sottogruppo degli eventi avversi in base all’età, al sesso e al fornitore dei farmaci in studio.
3.1.3. Fase di washout
I pazienti sono stati randomizzati dopo un periodo di washout di una settimana con placebo in singolo cieco [9]. Poiché 5 pazienti con fluoxetina contro 9 con placebo assumevano antidepressivi triciclici [9], il rischio di sintomi di astinenza era maggiore per i pazienti randomizzati al placebo.
3.1.4. Effetto della fluoxetina sulla depressione
Le analisi di efficacia hanno favorito la fluoxetina. Dopo 4 settimane, 6 pazienti avevano interrotto l’assunzione di fluoxetina e 12 quella di placebo [9]. Poiché la maggior parte delle analisi ha utilizzato il metodo dell’ultima osservazione riportata (LOCF), un numero maggiore di pazienti con placebo rispetto a quelli con fluoxetina ha riportato punteggi di depressione elevati.
Dopo 8 settimane, la differenza dal basale nel CDRS-R era maggiore di 9,7 con la fluoxetina rispetto al placebo utilizzando il metodo LOCF. Era di 2,5 utilizzando casi osservati su un grafico e di 6,0 su un altro grafico nella stessa pagina, senza alcuna spiegazione per questa discrepanza.
Nell’articolo pubblicato sono stati omessi i risultati relativi a BID e FGAS. Il CSR ha omesso entrambi i risultati e quelli relativi a WSAS e CGAS, affermando che le scale self-report non sono state raccolte in modo coerente (ma non fornendo alcuna prova in merito) e perché “i dati provengono da scale relativamente non validate”.
Lilly non ha spiegato i risultati discrepanti per la BPRS-C nel CSR e nella pubblicazione, ad esempio per quanto riguarda la fluoxetina, il punteggio medio dopo il trattamento è stato rispettivamente di 38,9 e 18,0, e anche i valori P erano nettamente diversi, P = 0,32 (calcolato da noi) contro P = 0,09 (Tabella 1).
L’articolo pubblicato combinava i risultati per le scale CDI e BDI, ma tutte le medie erano più piccole delle medie ponderate per le scale CDI e BDI nel CSR (vedi Tabella 1), il che è un’impossibilità matematica.
Nonostante il bias introdotto dall’uso dei valori di uscita, non sono state riscontrate differenze significative nella BPRS-C (P = 0,32), nella CGAS (P = 0,18) o negli esiti riportati dai pazienti, CDI/BDI (P = 0,58) e WSAS (P = 0,17) (Tabella 1) [7]. Emslie et al. hanno affermato che, “data l’ampia variabilità delle autosegnalazioni iniziali dei bambini, questi risultati sono difficili da interpretare [7]”. Tuttavia, i coefficienti di variazione (deviazioni standard divise per le medie) erano più piccoli per il BPRS-C e il CGAS che per il CDRS-R (Tabella 1).
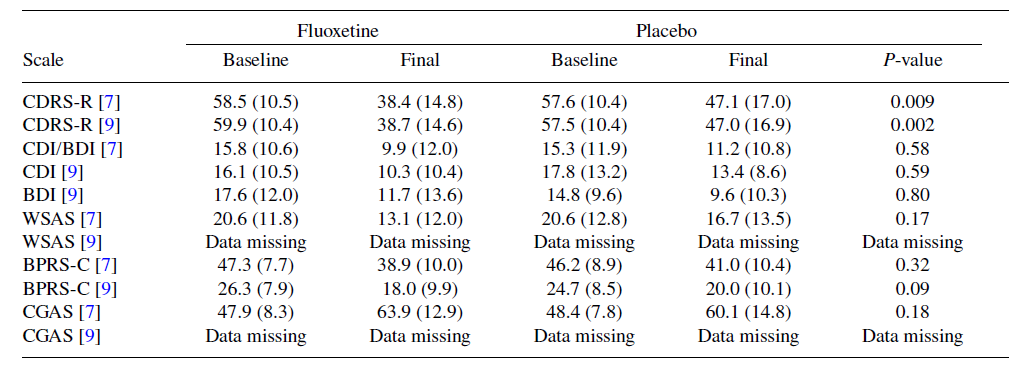
Discrepanze tra i risultati riportati nelle tabelle del rapporto pubblicato [7] e nel rapporto dello studio [9] per lo studio X065. P-valori per i dati pubblicati calcolati da noi sulla base dei punteggi finali; altri P-valori calcolati da Lilly sulla base delle variazioni dal basale (SD)
Il CSR ha fornito i valori di P per tutti i singoli 21 item del BPRS-C e ha sottolineato che la fluoxetina aumenta l’iperattività in modo significativo (P < 0,001) [9]. L’iperattività è un segnale d’allarme per il suicidio e la violenza [2], ma Lilly ha introdotto il concetto opposto: “Una diminuzione dell’ipoattività… che è associata al ritorno dei pazienti al normale funzionamento a tassi maggiori rispetto ai pazienti trattati con placebo”, trasformando un risultato potenzialmente dannoso in un beneficio. Non c’erano dati a sostegno di questa interpretazione e Emslie et al. hanno dichiarato che: “Non è noto se il trattamento a lungo termine comporti un miglioramento scolastico, del funzionamento generale o delle comorbidità concomitanti [7]”.
Nessun aggiustamento statistico può sostituire in modo affidabile i dati mancanti. Ci siamo quindi concentrati sui pazienti con sintomi minimi dopo 8 settimane. Solo 15 contro 11 pazienti avevano sintomi minimi (P = 0,49, secondo i nostri calcoli), definiti da Lilly come CDRS-R ≤ 28, e solo 14 contro 9 erano guariti (P = 0,34), definiti come CDRS-R ≤ 28 e un punteggio di miglioramento CGI di 1 o 2 [9]. Anche questi risultati possono essere falsati a favore della fluoxetina, poiché la remissione spontanea è comune e mancano più dati per i pazienti che assumono il placebo rispetto a quelli che assumono la fluoxetina (23 contro 15 hanno abbandonato il trattamento alla settimana 8).
3.1.5. Eventi avversi gravi e interruzioni
Le informazioni sulle interruzioni e sui tentativi di suicidio non sono coerenti. Nel rapporto pubblicato [7], 36 pazienti hanno interrotto il trattamento, nel CSR 38 [9]. Il numero, i motivi e la tempistica erano gli stessi solo in 5 delle 28 celle con informazioni (Tabella 2). Non è stata fornita alcuna spiegazione per queste discrepanze [9].
Nessun paziente è morto. Due pazienti hanno tentato il suicidio con la fluoxetina, rispettivamente dopo 12 e 15 giorni [9]. Questi tentativi di suicidio sono stati omessi dal rapporto pubblicato [7]. Uno di questi ha portato all’interruzione dello studio dopo 15 giorni e sembra che Emslie et al. l’abbiano definito una “violazione del protocollo” (vedi Tabella 2). Gli eventi avversi definitivamente o possibilmente predisponenti al suicidio in questo paziente secondo i nostri criteri sono stati reazione maniacale, insonnia e nervosismo (forse acatisia) [9]. Gli eventi predisponenti per l’altro paziente erano ansia, depressione, nevrosi, pensieri anormali, astenia e anche ipercinesia (caratteristiche dell’acatisia).
Altri quattro pazienti hanno interrotto la fluoxetina a causa di eventi avversi definiti “minimi” nel rapporto pubblicato, anche se tre di loro hanno sviluppato sintomi maniacali e il quarto ha avuto una grave crisi rash [7].
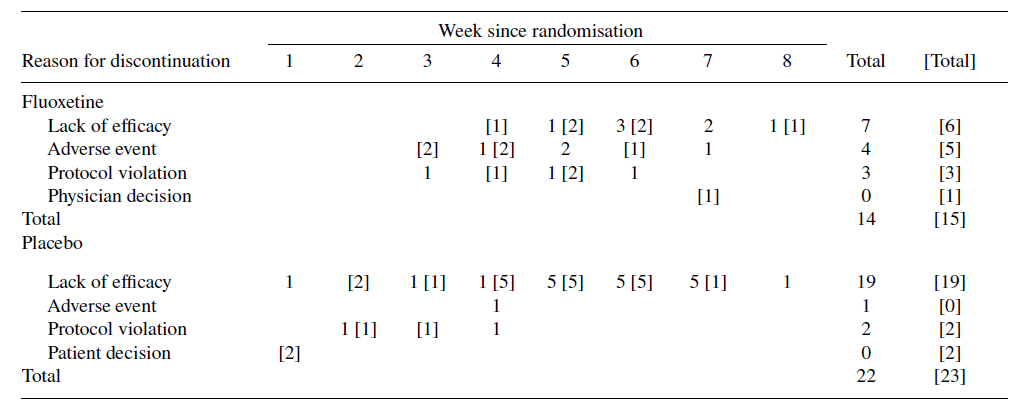
Pazienti interrotti nell’articolo pubblicato rispetto al CSR (numeri tra parentesi quadre) per lo studio X065
Il riassunto della RSI descriveva due pazienti con ipomania, uno con aumento dell’impulsività e uno con eruzione cutanea [9]. I racconti di questi pazienti erano estremamente brevi, tra le 17 e le 35 parole [9]. Uno aveva anche un attacco d’ansia e l’altro era affetto da ipercinesia. Il paziente con aumento dell’impulsività presentava ipercinesia, ansia e depressione, ma è stato codificato come disturbo di personalità.
Il CSR affermava che “Nessun paziente trattato con placebo ha interrotto il trattamento a causa di eventi avversi”, mentre l’articolo pubblicato menziona uno di questi pazienti, ma non l’evento che ha causato l’interruzione. Nel 2004, Emslie et al. hanno pubblicato ulteriori dati, menzionando questi due tentativi di suicidio ma attribuendone uno al gruppo placebo, con il riferimento “Emslie, comunicazione personale [12]”. Hanno inoltre affermato che un altro paziente con placebo ha interrotto il trattamento a causa della mania, anche se aveva ricevuto fluoxetina.
Il CSR ha registrato un maggior numero di pazienti che hanno abbandonato il placebo rispetto alla fluoxetina per mancanza di effetto (19 contro 6, P = 0,005), ma un minor numero di pazienti che hanno abbandonato per eventi avversi (0 contro 5, P = 0,056). Per ogni paziente è stata assegnata una sola causa [7,9]. Sommando entrambe le cause, 19 contro 11 pazienti si sono ritirati (P = 0,12, secondo i nostri calcoli). I farmaci concomitanti con proprietà sedative, spesso utilizzati per gestire gli eventi avversi legati al suicidio e alla violenza, sono stati menzionati per 11 pazienti in terapia con fluoxetina e per 3 pazienti in terapia con placebo.
3.1.6. Altri eventi avversi
Un paziente in trattamento con fluoxetina e tre con placebo non hanno manifestato nessuno dei 32 eventi avversi elencati nella Side-Effects Checklist [9]. I dati sugli eventi avversi non sollecitati e sugli eventi della lista di controllo degli effetti collaterali della fluoxetina non sono stati inclusi nel testo principale della RSI, ma sono stati inseriti dopo “Discussione e conclusioni generali”, nella sezione “Tabelle, figure e grafici non inclusi in altre sezioni [9]”. Nella tabella degli eventi avversi non richiesti, Lilly ha fornito 29 valori P e ha scritto che non c’erano differenze statisticamente significative, ma senza notare che un numero maggiore di pazienti con fluoxetina rispetto al placebo ha sperimentato uno o più eventi (P = 0,051, il valore P di Lilly) [9].
Al contrario, una tabella che riportava 28 delle 30 voci elencate nella lista di controllo degli effetti collaterali della fluoxetina non presentava un singolo valore P, anche se c’erano notevoli differenze. Ad esempio, 32 pazienti con fluoxetina contro 18 con placebo hanno sperimentato almeno un evento avverso (P = 0,008), 19 contro 6 hanno sperimentato irrequietezza (P = 0,005), 9 contro 1 hanno avuto incubi (P = 0,02) e 7 contro 4 si sono sentiti tesi all’interno. L’irrequietezza, compresa la sensazione di tensione interna, e gli incubi aumentano il rischio di suicidio e di violenza [4,5]. Lilly ha sostenuto che questa lista di controllo non è stata utilizzata in modo coerente nel corso dello studio e non è stata somministrata a tutti i pazienti randomizzati, ma non ha fornito informazioni sul numero di valori mancanti. Non sono stati forniti dati sulla gravità degli eventi avversi, anche se è stata valutata [9].
L’articolo pubblicato non presentava alcun dato ottenuto in uno dei tre modi appena descritti. Solo 49 delle 1099 parole (4%) della sezione Risultati erano relative alla sicurezza e riguardavano solo i pazienti interrotti [7]. Il rapporto pubblicato afferma che gli elettrocardiogrammi sono stati effettuati al basale e dopo 4 e 8 settimane, ma secondo l’RSI solo al basale e “non sono stati analizzati statisticamente”. I dati non sono stati riportati né nell’articolo pubblicato né nella RSI.
3.2. Risultati dello studio HCJE
HCJE è uno studio multicentrico condotto da Eli Lilly negli Stati Uniti dal 27 aprile 1998 al 16 dicembre 1999 [10]. Lo studio ha incluso 219 pazienti ambulatoriali (108 femmine) di età compresa tra gli 8 e i 17 anni (media 12,7) con disturbo depressivo maggiore, con una durata media dell’episodio di 61 settimane e un punteggio superiore a 40 sulla CDRS-R e un punteggio almeno moderato sulla scala CGI-Severity.
I pazienti sono stati trattati per una settimana con fluoxetina 10 mg una volta al giorno (109 pazienti) o placebo (110 pazienti), seguiti da 8 settimane con 20 mg o placebo (fase di trattamento acuto) e da altre 10 settimane in cui i non rispondenti a 20 mg di fluoxetina sono stati randomizzati a 20 mg o 40 mg di fluoxetina (che poteva essere aumentata a 60 mg 4 settimane dopo), mentre i restanti pazienti hanno continuato a somministrare 20 mg di fluoxetina o placebo. La terminologia era incoerente. Le 19 settimane sono state definite “fase di trattamento subcronico” anche se includevano la fase acuta, e il protocollo menzionava una fase di trattamento acuto di 19 settimane e una successiva fase di prevenzione delle ricadute di 32 settimane in cui i pazienti con un CDRS-R ≤ 28 a 19 settimane potevano essere randomizzati per continuare il trattamento attuale o passare al placebo.
3.2.1. Randomizzazione e cecità
La randomizzazione è stata stratificata per sesso ed età utilizzando una sequenza di randomizzazione generata dal computer. Non sono state fornite informazioni sulla dimensione del blocco. Erano disponibili codici sigillati, il che rischia di violare l’occultamento dell’assegnazione.
Il farmaco è stato confezionato in blister con un numero di confezione. Tutti i pazienti hanno ricevuto tre capsule al giorno contenenti 10 o 20 mg di fluoxetina o placebo abbinato. I pazienti che non tolleravano la fluoxetina potevano ridurre la dose da 20 a 10 mg o da 40 a 20 mg.
Non è chiaro se l’accecamento sia stato mantenuto dopo le prime 9 settimane, quando i pazienti che assumevano placebo e quelli che rispondevano a 20 mg di fluoxetina continuavano con il loro farmaco, mentre i non rispondenti alla fluoxetina venivano randomizzati a 20 o 40 mg di fluoxetina.
“Un numero minimo di personale Lilly” avrebbe visto la tabella di randomizzazione e i codici prima del completamento dello studio. Questo numero minimo comprendeva statistici, scienziati regolatori, analisti di sistemi, persone che lavoravano con la farmacocinetica, personale di medicina di laboratorio clinica e redattori medici, tutti con accesso non cieco ai dati durante lo studio. I pazienti che hanno abbandonato lo studio hanno potuto ricevere una terapia di salvataggio da un medico che non era in cieco, ma che “accettava di mantenere lo studio in cieco rispetto al personale dello studio”. Lilly ha riferito all’FDA che nella revisione dei registri delle fonti non era raro vedere annotazioni che definivano il trattamento in cieco del paziente o, in alcuni casi, i risultati della concentrazione plasmatica di fluoxetina [13].
3.2.2. Risultati
Tutti gli obiettivi dello studio erano incentrati sull’efficacia; nessuno sulla sicurezza. L’esito primario, CDRS-R, è stato dicotomizzato in una riduzione di almeno il 30% rispetto al basale. Sono stati descritti numerosi obiettivi secondari, esiti e confronti e ciò che è stato riportato non sempre corrisponde a quanto pianificato.
Nel protocollo, gli obiettivi secondari “sono i seguenti”, mentre nella RSI sono “inclusi i seguenti”, suggerendo che avrebbero potuto essere più di quelli elencati, come in effetti è stato. I tassi di remissione CDRS-R e i tassi di risposta CGI erano “analisi aggiuntive” esplorative nel protocollo e sarebbero state “condotte come ritenuto appropriato”, ma nel CSR erano “anche analizzate”, termine che Lilly utilizzava per le analisi pre-pianificate. Pertanto, le analisi esplorative definite nel protocollo hanno acquisito lo status di esiti secondari nel CSR.
Le scale di valutazione e le analisi sono state modificate. Il punteggio medio nel protocollo è diventato la variazione media per sette risultati nel CSR e in un altro punto del protocollo, e le analisi di sottogruppo per età sono diventate analisi di sottogruppo per età, sesso e storia familiare di depressione.
I risultati di efficacia sono stati:
(1) Variazione media della CDRS-R
(2) “Valutazione del CDRS-R
(3) CGI-Severità
(4) CGI-Miglioramento
(5) Scala di valutazione della depressione Montgomery-Asberg (MADRS)
(6) BDI
(7) CDI
(8) Valutazione globale del funzionamento (GAF)
(9) Funzionamento attuale
(10) Indice CGI-Efficacy
(11) Hamilton Anxiety Rating Scale (non menzionato nel protocollo modificato)
(12) Kiddie Schedule for Affective Disorders and Schizophrenia-Present and Lifetime (K-SADSPL) (solo per il modulo dei Disturbi Affettivi), che nel protocollo era Kiddie Schedule for Affective Disorders and Schizophrenia (K-SADS).
Gli esiti clinici sono stati registrati a ogni singola visita dopo la randomizzazione (11 volte), mentre gli esiti valutati dai pazienti (CDI o BDI, a seconda dell’età) sono stati registrati solo a 5 visite.
Gli eventi avversi non richiesti sono stati raccolti interrogando i pazienti sulla presenza di eventi avversi “in modo non diretto”, senza ulteriori istruzioni. Un modulo per la registrazione di tutti gli eventi avversi verificatisi dall’ultima visita e di tutte le anomalie clinicamente rilevanti riscontrate all’esame fisico o all’ECG occupava una pagina. Aveva un riquadro di 4,5 cm2 per ogni descrizione e tre codici di gravità (lieve, moderata e grave) che non erano definiti da nessuna parte. I moduli per i benefici occupavano 10 pagine. C’era un modulo opzionale di una pagina per i commenti sui benefici o sui valori di laboratorio aberranti, ma non sugli eventi avversi.
Gli eventi avversi richiesti sono stati rilevati con la Side-Effects Checklist. I termini verbatim sono stati codificati come termini COSTART da personale Lilly in cieco. Non è stata utilizzata la lista di controllo degli effetti collaterali della fluoxetina sviluppata da Lilly e utilizzata nello studio X065.
Sono stati misurati pressione sanguigna, frequenza cardiaca, altezza e peso. Il testo riporta che gli ECG sono stati eseguiti in vari momenti dello studio. Il calendario degli eventi indica che sono stati effettuati solo alla prima visita e dopo 19 settimane, ma una tabella delle misurazioni mancanti mostra che sono mancate solo 41 registrazioni, in varie visite numerate durante l’intero studio.
3.2.3. Analisi statistiche
Sono state effettuate numerose analisi che non erano state prestabilite nel protocollo o nel piano di analisi statistica, anche se quest’ultimo è stato aggiornato il 7 febbraio 2000, due mesi dopo la conclusione dello studio di 19 settimane, quando molti dipendenti Lilly avevano visto i valori non ciechi.
Abbiamo riscontrato un uso problematico delle statistiche e informazioni contraddittorie. È stato dichiarato che tutti i pazienti con almeno una visita successiva alla randomizzazione sarebbero stati inclusi nelle analisi di efficacia, ma questo non valeva per le analisi primarie di efficacia per le quali erano necessarie due visite, tra cui “almeno una settimana di assunzione di fluoxetina 20 mg/die”.
Contrariamente a quanto dichiarato dall’azienda di aver condotto analisi intention-to-treat, alcuni pazienti con visite successive alla randomizzazione sono stati esclusi e i pazienti che sono stati interrotti non sono stati valutati alla fine dello studio. Poiché era stato dichiarato che i pazienti sarebbero rimasti nei gruppi a cui erano stati assegnati casualmente anche se non avessero seguito il protocollo, non aveva senso interrompere i pazienti, il cui unico effetto era quello di falsare i risultati dello studio a favore della fluoxetina.
Le analisi della RSI includevano:
(1) riduzione del 10%, 20%, 30%, 40%, 50%, 60% e 70% del CDRS-R (solo il 30% era prespecializzato nel protocollo)
(2) variazione della CDRS-R dal basale a ogni visita successiva
(3) Tasso di remissione del CDRS-R
(4) Analisi dei casi osservati
(5) ANOVA su CDRS-R
(6) Variazione del CDRS-R subtotale
(7) Variazione della CGI-Severità
(8) Variazione del BDI
(9) Variazione del CDI
(10) Variazione di MADRS
(11) Variazione di HAMA
(12) Variazione di GAF
(13) Miglioramento della CGI
(14) ANOVA sui valori endpoint
(15) ANOVA sui cambiamenti
(16) ANOVA con interazione tra trattamento, sperimentatore, sesso, fascia d’età e trattamento per sperimentatore
(17) ANOVA utilizzando un approccio a modello misto con le variabili dipendenti rappresentate dal CDRS-R al basale e al post-basale e i fattori indipendenti rappresentati da trattamento, sperimentatore, interazione trattamento-investigatore, visita e interazione trattamento-visita
(18) Tasso di risposta CGI-definito
(19) Recupero
(20) Percentuale di pazienti con una diagnosi di depressione utilizzando il K-SADS-PL all’endpoint.
(21) Analisi sia sui dati originali che su quelli trasformati per rango
(22) Modelli di regressione logistica con trattamento, sperimentatore, sesso, categoria di età e interazione trattamento-sperimentatore per il confronto delle percentuali.
Più avanti nella RSI sono state effettuate ulteriori analisi, ad esempio per 4 subtotali della CDRS-R e per ciascuno dei 17 singoli item. Sono state eseguite analisi di sottogruppo sulla risposta e sulla variazione media della CDRS-R; sul miglioramento della CGI-; sugli eventi avversi non sollecitati e sollecitati, separatamente per bambini e adolescenti, maschi e femmine, e per pazienti con e senza una storia familiare di depressione. Sono stati inoltre presentati i risultati, con i valori P, per ciascuno dei 15 siti di sperimentazione per CDRS-R, CGI-Miglioramento e MADRS.
Inoltre, “dopo aver esaminato alcuni dei risultati di questo studio, sono state eseguite ulteriori analisi esplorative. Sono state effettuate ulteriori analisi di sottogruppo e analisi “complete” dei dati di laboratorio, ECG e segni vitali… Una seconda serie di letture ECG in cieco eseguite da un cardiologo pediatrico ha integrato le letture originali”.
Anche se solo il 34% dei pazienti ha completato tutte le 19 settimane, le analisi dopo 19 settimane sono state chiamate analisi intermedie. Tutte le analisi effettuate dopo 9 settimane sono state ripetute dopo 19 settimane insieme ad altre, ad esempio includendo i pazienti che avevano ricevuto “almeno 4 settimane di fluoxetina 20 mg/die”. Poiché non viene detto nulla sul gruppo placebo, non è chiaro se gli stessi criteri siano stati applicati al gruppo placebo o se tutti i pazienti placebo siano stati inclusi, indipendentemente dall’assunzione di placebo.
Un’appendice di due pagine sui “pazienti esclusi dall’analisi di efficacia” è apparsa 90 pagine dopo la conclusione della sezione sull’analisi statistica, a pagina 2356. Si trattava di una tabella senza testo esplicativo che elencava 9 pazienti in trattamento con placebo, con il loro numero di paziente e i valori di CDRSTL17.
I valori CDRSTL17 erano probabilmente i punteggi CDRS-R al basale, poiché il numero di visita corrispondeva alla visita di randomizzazione per tutti i 9 pazienti. Tuttavia, la tabella indicava che tutti e 9 i pazienti erano stati esclusi alla visita di randomizzazione, mentre era vero solo per uno di loro. Nel rapporto sono riportate informazioni sparse che indicano che 3 dei pazienti sono stati sospesi dopo 7-10 giorni. I restanti 5 pazienti non sono stati interrotti. Secondo il protocollo e il CSR, solo 4 pazienti con placebo avrebbero dovuto essere esclusi dall’analisi di efficacia primaria, ma altri 5 sono stati esclusi, senza spiegarne il motivo, mentre nessun paziente con fluoxetina è stato escluso. La differenza tra 0 e 9 esclusioni è altamente improbabile che si sia verificata per caso (P = 0,003, secondo i nostri calcoli).
3.2.4. Fase di washout
Era previsto un periodo di valutazione diagnostica di due settimane a partire dalla settimana-3 (chiamato anche periodo “senza farmaci” in una figura), ma non era spiegato se il precedente trattamento antidepressivo fosse stato interrotto o se non fossero ammessi nuovi farmaci. In una tabella di violazioni del protocollo di 2405 pagine dopo, un paziente ha ricevuto l’ultima dose di sertralina 12 giorni prima della prima visita, che avrebbe dovuto essere 14 giorni prima. Solo in questa tabella si è potuto notare che doveva essere obbligatorio sospendere gli antidepressivi precedenti 5 settimane prima della randomizzazione.
Un periodo di washout in singolo cieco di una settimana con placebo è iniziato alla settimana-1, ma la settimana-3 è stata la linea di base per valutare la cosiddetta risposta al placebo alla settimana 0. Questo non ha senso perché il placebo è stato somministrato in un’unica settimana. Questo non ha senso perché il placebo non è stato somministrato prima della settimana-1.
Gli antidepressivi precedenti sono stati elencati 6 volte per la fluoxetina e 6 per il placebo, ma poiché i pazienti, i farmaci e gli eventi avversi non sono stati collegati, è stato impossibile sapere se alcuni degli eventi avversi gravi nel periodo introduttivo di tre settimane fossero effetti iatrogeni da sospensione. Gli eventi avversi gravi si sono verificati in tre pazienti non randomizzati: Alla settimana -3, un paziente presentava aggressività esplosiva e un altro sintomi psicotici; il terzo paziente aveva un’ideazione suicida alla settimana -1 codificata come depressione.
3.2.5. Farmaci concomitanti
A giudicare dai moduli di refertazione clinica, ai pazienti non è stato chiesto di routine quali farmaci stessero assumendo. Dopo 447 pagine di moduli di report in bianco, c’era un modulo sui farmaci concomitanti che gli sperimentatori potevano utilizzare “all’ingresso e durante lo studio”, ma poiché non era obbligatorio utilizzarlo, le informazioni sugli altri farmaci utilizzati durante lo studio non erano affidabili. I farmaci concomitanti sono stati utilizzati da un maggior numero di pazienti con fluoxetina rispetto al placebo, l’82% contro il 66% durante le prime 9 settimane (P = 0,01) e l’84% contro il 72% durante tutte le 19 settimane (P = 0,03). Il paracetamolo è stato usato più spesso (P = 0,02 e 0,04, rispettivamente). Per i farmaci con proprietà sedative, le occorrenze sono state: antistaminici 38 contro 31; sedativi/ipnotici 2 contro 5; antipsicotici 1 contro 0.
3.2.6. Effetto della fluoxetina sulla depressione
Come nello studio X065, tutte le analisi di efficacia sono state falsate a favore della fluoxetina perché il grado di depressione dopo 9 e 19 settimane non era noto per i pazienti interrotti. Dopo due settimane, nessuno aveva abbandonato il trattamento con la fluoxetina contro i 10 del placebo. La maggior parte delle analisi ha utilizzato il metodo LOCF, ma Lilly non ha avvertito i suoi lettori della distorsione che ne derivava.
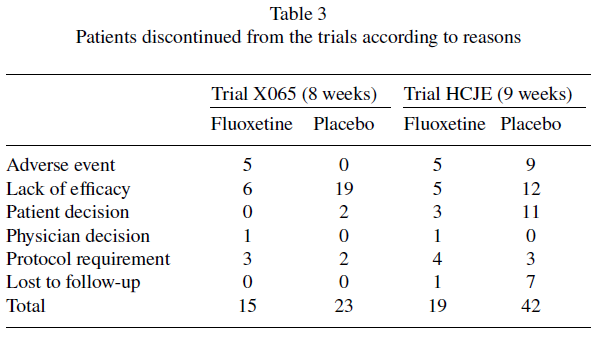
Il revisore medico dell’FDA ha notato il numero considerevolmente maggiore di abbandoni con il placebo rispetto alla fluoxetina e che il modello era “piuttosto insolito”, perché c’erano più abbandoni con il placebo che con la fluoxetina per eventi avversi (9 contro 5), decisione del paziente (11 contro 3) e perdita del follow-up (7 contro 1) [13]. Lo studio X065 ha registrato 0 abbandoni per eventi avversi con il placebo contro 5 con la fluoxetina, e non ci sono state perdite al follow-up (vedere Tabella 3). La FDA ha osservato che la Lilly aveva fornito dichiarazioni secondo cui i suoi ricercatori clinici non avevano ricevuto il pagamento in cambio di risultati particolari [13].
Molte tabelle e grafici erano confusi, con il tipo di analisi non specificato e la terminologia poco chiara, ad esempio “dal basale all’endpoint” poteva significare casi osservati o LOCF. Il risultato primario è stato descritto come: “È stata osservata una forte tendenza numerica, con 71 (65%) pazienti trattati con fluoxetina che hanno soddisfatto i criteri di risposta rispetto a 54 (54%) pazienti trattati con placebo;
Tuttavia, la differenza tra i gruppi di trattamento non era statisticamente significativa (P = 0,093) per questo periodo di trattamento di 9 settimane”. Questa analisi non ha prodotto risultati per il 28% dei pazienti che hanno interrotto il trattamento.
La differenza nei punteggi di cambiamento del CDRS-R dopo 9 settimane era di 5,9 in un grafico, mentre era di 7,2 in una tabella. In un altro grafico, la differenza era di 7,2 usando la LOCF, il che suggerisce che Lilly ha usato la LOCF anche per la tabella senza dirlo.
Nonostante le figure, il placebo tendeva a essere migliore della fluoxetina secondo la valutazione dei pazienti: “I pazienti trattati con placebo hanno mostrato maggiori riduzioni numeriche nella variazione dal basale dei punteggi totali di CDI e BDI rispetto ai pazienti trattati con fluoxetina. La differenza tra i gruppi di trattamento non era statisticamente significativa”.
Nell’articolo pubblicato, la mancanza di un effetto della fluoxetina su CDI e BDI è stata menzionata in questi termini: “Data l’alta percentuale di pazienti in questo studio che avevano una comorbilità con l’ADHD, potrebbe non essere sorprendente che i risultati delle misure valutate dal clinico non siano stati riflessi dai risultati delle scale valutate dal paziente [8]”. Tuttavia, solo il 14% dei bambini aveva una diagnosi di ADHD, e non è probabile che tale diagnosi renda inaffidabile la valutazione degli effetti dei farmaci da parte dei bambini.
Il rapporto terminava a pagina 224, ma era seguito da altre 2325 pagine con dati aggiuntivi. Gli psichiatri hanno utilizzato un indice CGI-Efficacy con 8 categorie per “valutare l’effetto terapeutico complessivo insieme agli effetti collaterali per ciascun paziente”. Hanno valutato per ogni paziente se il miglioramento della depressione fosse superiore agli eventuali danni del farmaco in termini di interferenza con le attività quotidiane (che non è stata definita). Lilly ha sostenuto che i risultati indicavano che gli effetti terapeutici erano superiori agli effetti collaterali, poiché il 58% contro il 40% aveva un punteggio favorevole. Abbiamo combinato i dati delle 8 categorie sottraendo gli esiti negativi da quelli positivi e abbiamo scoperto che il 59% contro il 55% aveva un esito positivo (P = 0,58).
La fluoxetina è risultata inefficace dopo 19 settimane per la CDRS-R quando sono stati utilizzati casi osservati (la differenza rispetto al placebo è stata di 2,5, P = 0,36). Per l’analisi LOCF, la differenza era significativa per tutte le visite, anche dopo la prima settimana, quando i pazienti con fluoxetina avevano ricevuto solo 10 mg, ma dopo 19 settimane, la differenza era solo di 4,8 (P = 0,02). In un’ANOVA dei cambiamenti, la differenza era di 1,7 (P = 0,13).
In molti casi, non siamo riusciti a capire come le analisi aggiuntive differissero da quelle precedenti e perché i numeri dei pazienti differissero quando avrebbero dovuto essere gli stessi. A differenza dei risultati a 9 settimane riportati nel riassunto del rapporto (vedi sotto), diversi risultati non erano più statisticamente significativi dopo 19 settimane, ad esempio per MADRS, CGI-Improvement, risposta basata su CGI-Improvement, recupero basato sui punteggi CDRS-R e CGI-Improvement e subtotale dell’umore CDRS-R. A titolo di esempio, per CGI-Improvement, P = 0,03 dopo 9 settimane e P = 0,32 dopo 19 settimane.
3.2.7. Eventi avversi gravi e interruzioni
C’erano narrazioni per il 10% dei pazienti (6 in X065 e 24 in HCJE per eventi post-randomizzazione). Come spieghiamo di seguito, questi erano molto brevi, poco chiari e incompleti. Si sono verificati errori importanti ed è stato necessario combinare due serie di tabelle separate da 668 pagine. Le CSR utilizzavano due serie di termini in modo intercambiabile, il termine letterale riportato dagli investigatori e il termine codificato aggiunto dalla società. Lilly non ha specificato quali termini sono stati usati in tre serie di tabelle molto diverse. In una tabella, l’intestazione della colonna era “Termine di classificazione dell’evento”, ma ciò che veniva riportato era il termine letterale.
Nessun paziente è morto. Due pazienti hanno avuto eventi avversi gravi con fluoxetina e quattro con placebo, ma la segnalazione è stata breve, opaca e contraddittoria ei termini non erano gli stessi nelle tabelle che descrivevano gli stessi pazienti. Combinando tre tabelle e una figura è risultato che un paziente in trattamento con fluoxetina ha sviluppato un’idea suicidaria 89 giorni prima della prima visita ed è stato interrotto 70 giorni dopo la randomizzazione quando il suicidio era diventato grave e il paziente è stato ricoverato in ospedale. L’investigatore ha ritenuto che l’evento fosse correlato al farmaco in studio, ma Lilly lo ha codificato come depressione. La tendenza suicidaria è diventata grave due settimane dopo che alcuni pazienti erano stati nuovamente randomizzati a 40 mg di fluoxetina, ma non è stato possibile valutarne la possibile relazione con l’aumento della dose, poiché non c’erano informazioni sulla dose. Una delle tabelle indicava che questo paziente era stato ritirato alla randomizzazione e che la fluoxetina non poteva quindi aver causato il suicidio.
L’altro paziente in terapia con fluoxetina aveva tonsille gonfie codificate come faringite ed era stato sottoposto a tonsillectomia. La narrazione ha rivelato che il paziente soffriva di molto altro, tra cui stanchezza e irritabilità. I quattro eventi avversi gravi del placebo erano infezione renale, dolore/appendicite addominale, comportamento aggressivo codificato come ostilità e comportamento automutilatorio codificato come lesione intenzionale. La narrazione per il paziente con appendicite utilizzava il termine infezione virale codificata come infezione, il che creava confusione, poiché questi dati provenivano dalla fase di prevenzione delle ricadute dello studio anche se la società ha specificato in numerosi punti che i dati di quella fase non sarebbero stati inclusi in la CSR.
Il paziente con comportamento aggressivo e il paziente con comportamento automutilatorio presentavano sintomi alla prima visita e sono stati sospesi rispettivamente 10 e 37 giorni dopo la randomizzazione, quando i sintomi erano diventati gravi, e sono stati ricoverati in ospedale. Non avevamo informazioni che ci permettessero di giudicare se gli eventi potessero essere effetti iatrogeni causati dalla sospensione di un antidepressivo prima della randomizzazione.
Le informazioni sul paziente con comportamento aggressivo apparivano in una tabella, che mescolava pazienti non randomizzati con pazienti con eventi avversi gravi e pazienti ritirati a causa di eventi avversi non gravi. L’intestazione della tabella era comunque “Tutti i pazienti randomizzati”. Poiché non c’erano date, il paziente potrebbe non essere stato randomizzato.
Quando il comportamento automutilatorio è codificato come danno intenzionale, nasconde se si tratta di violenza nei confronti di sé o degli altri. Un breve racconto di 65 parole ha rivelato che questo paziente aveva sia un’ideazione suicidaria che un’ideazione omicida (codificata come ostilità) ed è stato ricoverato in ospedale per questi motivi.
Durante le prime 9 settimane, 19 contro 42 pazienti hanno interrotto lo studio, ma il rapporto di Lilly al Data Monitoring Board due mesi dopo il completamento dello studio ha descritto 15 contro 33 interruzioni. Questi numeri risalgono alla settimana 7 senza una spiegazione del motivo per cui il Consiglio non ha ottenuto i risultati completi. Questo errore è stato ripetuto per i risultati delle 19 settimane.
Undici pazienti in ciascun gruppo hanno interrotto il trattamento a causa di eventi avversi, sei dei quali per motivi psichiatrici non gravi trattati con fluoxetina e uno con placebo. I termini per la fluoxetina erano agitazione (dopo 28 giorni), umore elevato codificato come euforia (65 giorni), aggressività fisica codificata come ostilità (98 giorni), iperattività codificata come ipercinesia (18 giorni), mania (32 giorni) e disinibizione comportamentale codificato come disturbo di personalità (73 giorni).
Secondo le narrazioni, questi pazienti erano più gravemente colpiti di quanto suggerissero le tabelle. L’agitazione era grave e ulteriori eventi includevano irritabilità, affaticamento, diminuzione della concentrazione, insonnia, rabbia, sentire le voci, pensieri in corsa, sbalzi d’umore e capricci d’ira, che suggerivano psicosi indotta da farmaci e forse acatisia.
Il paziente con euforia è stato interrotto a causa di umore elevato, maggiore irritabilità, irrequietezza, maggiore iperattività e impulsività, eloquio pressato, tangenzialità, fuga delle idee, belligeranza e lieve scioltezza delle associazioni. Questi eventi predispongono al suicidio e alla violenza, che non si potrebbe sospettare dal termine dello sperimentatore umore elevato o dal termine codificato euforia. Il paziente con iperattività aveva una diagnosi di ADHD. Dopo 22 giorni di fluoxetina, i sintomi dell’ADHD erano diventati estremi, il che potrebbe essere un’acatisia indotta da farmaci. Il paziente con mania presentava anche irritabilità, agitazione, insonnia, linguaggio pressato e deliri.
Un settimo paziente ha interrotto la fluoxetina dopo 84 giorni a causa di iperplasia endometriale. La narrazione di 167 parole riguardava solo questa diagnosi insolita in una ragazza di 15 anni, ma l’elenco dei sintomi includeva acatisia.
Un ottavo paziente ha interrotto la fluoxetina dopo 120 giorni a causa di un aumento della gravità dell’emicrania. Sogni brutti e aggressività si sono verificati 1-2 giorni prima della randomizzazione durante il washout del placebo. Durante il trattamento si sono verificati disturbi del sonno, tremori, brutti sogni e irrequietezza intermittente codificata come acatisia.
Un paziente in trattamento con placebo aveva ansia alla randomizzazione e ha interrotto 7 giorni dopo. Due giorni dopo la randomizzazione questo paziente ha sviluppato astenia e, dopo sette giorni, era “agitato e urlava contro sua madre. Il paziente ha affermato che il farmaco in studio lo rendeva nervoso, gli dava mal di testa e gli faceva male lo stomaco. Il paziente ha chiesto di essere ritirato dallo studio”. Questo paziente potrebbe aver sofferto di acatisia a causa della sospensione del precedente farmaco.
In totale, 7 pazienti con fluoxetina rispetto a 3 con placebo hanno manifestato eventi avversi psichiatrici che hanno portato all’interruzione, che diventano 9 rispetto a 3 pazienti con eventi psichiatrici significativi se si aggiungono i due pazienti con acatisia trattati con fluoxetina.
Le informazioni in un articolo pubblicato del 2004, che riguardava solo la sicurezza, erano in netto disaccordo con i dati nella CSR anche se tutti e cinque gli autori provenivano da Lilly [14]. L’articolo affermava che 49 pazienti contro 47 “hanno completato 19 settimane di trattamento”, ma i numeri corretti erano 40 contro 35. L’articolo rilevava che 4 pazienti in trattamento con fluoxetina hanno riportato un evento correlato al suicidio o all’autolesionismo, ma abbiamo trovato solo uno di questi pazienti nella CSR (che fu ricoverato in ospedale per ideazione suicidaria). Gli altri tre pazienti, due con ideazione suicidaria e uno con automutilazione, non sono stati descritti.
Il documento ha anche rilevato che 4 pazienti trattati con placebo hanno riportato un evento correlato al suicidio o all’autolesionismo, ma ne abbiamo trovato solo uno (che è stato ricoverato in ospedale per suicidio e automutilazione). Un paziente ha assunto 9 capsule di placebo invece di 3, uno ha fatto commenti sul desiderio di morire e il terzo paziente ha riferito di ideazione suicidaria.
Queste discrepanze suggeriscono che l’azienda ha accesso alle informazioni relative al suicidio escluse dalla CSR, che affermava che “non c’era differenza nel tasso di eventi correlati al suicidio segnalati tra fluoxetina e placebo [14]”. Ciò contraddiceva i nostri risultati (vedi Tabella Supplementare S1).
3.2.8. Altri eventi avversi
Quasi tutti i pazienti hanno manifestato un evento avverso richiesto, 105 trattati con fluoxetina contro 100 con placebo dopo 9 settimane (P = 0,17). I numeri dopo 19 settimane erano 107 contro 101 (P = 0,06) in un’analisi e 108 contro 102 (P = 0,04) in un’altra. Per gli eventi avversi non richiesti, dopo 9 settimane (P = 0,02) e 101 contro 87 dopo 19 settimane (P = 0,006) sono stati trattati 94 con fluoxetina contro 80 con placebo.
Dopo 9 settimane, più pazienti avevano avuto eventi a carico del sistema nervoso trattati con fluoxetina rispetto al placebo, 35 contro 24 (P = 0,095). Lilly non ha commentato questo, anche se il P = 0,093 per il risultato di efficacia primaria è stato chiamato “tendenza numerica forte”. Dopo 19 settimane, la differenza era significativa, 42 contro 28 pazienti (P = 0,01, numero necessario per nuocere 6). Ci sono stati anche più eventi relativi al sistema respiratorio, 61 contro 42 (P = 0,01) e ai sensi speciali, 16 contro 4 (P = 0,005). Non c’erano dati sulla gravità nella parte principale della CSR e Lilly non ha definito i tre gradi di gravità utilizzati.
Nelle sperimentazioni sui farmaci, le definizioni usuali sono:
(1) Lieve: consapevolezza del segno o del sintomo, ma facilmente tollerabile.
(2) Moderato: disagio tale da causare interferenze con le attività abituali.
(3) Grave: invalidante con incapacità di lavorare o svolgere attività abituale.
Per i farmaci che hanno solo effetti sintomatici, i danni che interferiscono con le attività abituali sono rilevanti per una valutazione dell’equilibrio tra benefici e danni. Se viene scelto un solo confronto, non dovrebbe quindi essere per eventi gravi, come ha fatto Lilly, ma per eventi classificati come moderati o gravi.
Lilly ha riportato 19 contro 15 pazienti con uno o più eventi avversi gravi dopo 9 settimane (P = 0,46), mentre abbiamo riscontrato 68 contro 56 con eventi moderati o gravi (P = 0,10). Dopo 19 settimane, Lilly ha affermato che non c’erano più pazienti con eventi gravi in fluoxetina rispetto al placebo, 22 contro 18, ignorando che 78 contro 64 pazienti avevano eventi avversi moderati o gravi (P = 0,047, il nostro calcolo, numero necessario per danneggiare 7 ).
Lilly ha minimizzato ancora di più gli eventi avversi sollecitati osservando che “non c’erano differenze clinicamente rilevanti nell’intensità massima per qualsiasi evento avverso richiesto emergente dal trattamento”. Tuttavia, utilizzando la terminologia di Lilly, c’era una tendenza “forte” per eventi avversi gravi dopo 9 settimane, 64 contro 52 (P = 0,105), che diventavano 71 contro 57 dopo 19 settimane (P = 0,055). Dopo 9 settimane, un numero significativamente maggiore di pazienti trattati con fluoxetina rispetto a quelli trattati con placebo si sentiva assonnato, 19 contro 7 (P = 0,01), con difficoltà ad andare d’accordo con i genitori, 19 contro 9 (P = 0,045) e difficoltà a prestare attenzione, 18 contro 7 ( P = 0,02). Lilly ha affermato che le differenze erano minime e che gli eventi avversi non hanno portato a interruzioni. Tuttavia, le differenze erano di circa il 10%, il che significa che per ogni 10 pazienti trattati con fluoxetina, uno era gravemente danneggiato. Nove contro 5 pazienti hanno avuto gravi problemi con la posizione immobile, cosa su cui Lilly non ha commentato sebbene potesse significare acatisia.
La fluoxetina ha ridotto gli aumenti di altezza e peso in 19 settimane rispettivamente di 1,0 cm e 1,1 kg (P = 0,008 per entrambi). Non c’erano dati su questi danni nell’articolo pubblicato [8] e nessun commento su di essi nella CSR quando sono stati presentati [10].
La ditta ha concluso che “fluoxetina da 20 a 60 mg/die è sicura”. Discutendo i dati a 19 settimane per tutti i pazienti, il CSR ha affermato che il significato dell’effetto sull’altezza era incerto, poiché studi precedenti non lo avevano trovato. Lilly ha ripetuto questa affermazione nel suo articolo pubblicato sulla sicurezza e ha aggiunto che “la fiducia nell’interpretazione di questo risultato è limitata, dato che l’altezza in questo studio non è stata raccolta in modo standardizzato. Inoltre, poiché le misurazioni sono state registrate e arrotondate al pollice più vicino, una piccola imprecisione nella misurazione potrebbe potenzialmente comportare una differenza di 1 pollice nell’altezza registrata [14]”. Tuttavia, se le registrazioni sono arrotondate, ciò ha effetto su entrambi i gruppi. Inoltre, la preoccupazione è in contrasto con il fatto che l’altezza di ciascun paziente è stata riportata con due decimali (es. 160,02 cm) e il peso con otto decimali (es. 47,62719885 kg) [14].
La fluoxetina ha aumentato l’intervallo QTc di 6,95 msec (P = 0,02 per la differenza rispetto al placebo). Dopo numerose analisi e manipolazioni su sei pagine, ad es. mostrando che il valore P non era statisticamente significativo per i bambini con intervalli ECG fuori range e non era presente nelle analisi dei sottogruppi per età, Lilly ha concluso che “Due analisi indipendenti e in cieco delle variazioni dell’intervallo ECG non hanno rivelato cambiamenti clinicamente significativi in qualsiasi parametro ECG”. Infine, la fluoxetina ha aumentato il colesterolo sierico (differenza rispetto al placebo 0,2 mmol/L, P = 0,01).
3.2.9. Ulteriori analisi di eventi avversi
In un “Riassunto dei dati sulla sicurezza” di 1160 pagine, per le variazioni di altezza e peso, Lilly ha introdotto un nuovo intervallo, tutti i pazienti randomizzati “che non hanno interrotto prima della Visita 11”. Questa visita è stata di due settimane nell’ultimo periodo di 10 settimane. Non c’era alcuna spiegazione del motivo per cui questo intervallo è stato scelto. I dati sugli ECG sono stati presentati anche utilizzando il nuovo cut-off di 11 settimane. Come prima, l’effetto dannoso della fluoxetina è stato rimosso dicotomizzando i dati e osservando i valori fuori range.
3.3. Le conclusioni di Lilly
Le CSR per entrambi gli studi sono iniziate con un riassunto di 3 pagine, che includeva una tabella senza dati ma con 9 e 13 valori P, rispettivamente, per i risultati di efficacia selezionati, che erano tutti statisticamente significativi [8,10]. Non vi era alcuna indicazione che fosse stato utilizzato LOCF.
Per lo studio HCJE, 7 dei 13 esiti non erano prespecificati nel protocollo: riduzioni del 20% e 40%, recupero e quattro punteggi subtotali sul CDRS-R, che abbiamo visto solo negli studi sulla fluoxetina. Il documento pubblicato non menzionava l’uso di LOCF o che alcuni dei risultati non erano prespecificati [8]. Sei degli otto autori del documento erano dipendenti di Lilly e “potrebbero possedere azioni di quella società”. I due autori rimanenti erano consulenti pagati per Lilly.
L’affermazione che dosi di 40 e 60 mg al giorno fossero più efficaci di 20 mg in coloro che non avevano risposto a 20 mg non era corretta.
Una successiva sezione “Discussione e conclusioni generali” di 2,5 pagine ha elogiato i benefici e l’assenza di danni della fluoxetina, senza menzionare che i bambini non hanno trovato la fluoxetina efficace o che il numero necessario per nuocere era solo 6 per eventi del sistema nervoso e 7 per eventi moderati o gravi eventi avversi. Contraddicendo i dati, ha affermato che non vi erano “cambiamenti clinicamente significativi in alcun parametro ECG” e che la valutazione di eventi avversi, segni vitali (che includevano altezza e peso) e dati di laboratorio hanno dimostrato la sicurezza di fluoxetina da 20 a 60 mg al giorno [10].
3.4. Eventi avversi che predispongono alla violenza contro se stessi o gli altri
Poiché non avevamo accesso ai dati dei singoli pazienti, abbiamo contato gli eventi anche se alcuni pazienti avevano più di un evento predisponente (vedere la tabella supplementare S1). Per gli eventi avversi sollecitati, 11 dei 32 eventi sono stati considerati precursori definiti o possibili del suicidio o della violenza, o entrambi, dal valutatore in cieco (DH), e 9 sono stati considerati precursori definitivi. C’erano 131 contro 124 precursori definiti o possibili nello studio X065; 396 contro 396 nello studio HCJE dopo 9 settimane; e 450 contro 440 dopo 19 settimane. Per i precursori definiti, i numeri erano 100 contro 100, 314 contro 320 e 358 contro 353, rispettivamente.
Per gli eventi avversi non richiesti, i dati erano molto diversi. C’erano 84 contro 72 precursori definiti o possibili nello studio X065; 72 contro 42 nello studio HCJE dopo 9 settimane; e 102 contro 49 dopo 19 settimane. C’erano rispettivamente 70 contro 61, 40 contro 18 e 58 contro 22 precursori definiti. Per la checklist degli effetti collaterali della fluoxetina, utilizzata solo nello studio X065, c’erano 58 contro 23 precursori definiti o possibili e 39 contro 13 precursori definiti.
Prendendo insieme i due studi, il verificarsi di eventi avversi che sicuramente predispongono alla violenza contro se stessi o gli altri che portano all’interruzione è stato di 11 contro 3.
Uno dei precursori più forti della violenza contro se stessi o gli altri è l’acatisia. In un’analisi esplorativa, abbiamo incluso l’acatisia e altri sintomi potenzialmente correlati (vedi Tabella Supplementare S2). Per gli eventi avversi non richiesti, si sono verificati 37 contro 32 di tali eventi avversi nello studio X065; 38 contro 16 nello studio HCJE dopo 9 settimane; e 51 contro 24 dopo tutte le 19 settimane. Abbiamo incluso il nervosismo perché Lilly aveva codificato l’irrequietezza come nervosismo in una narrazione paziente. Per la checklist degli effetti collaterali della fluoxetina, ci sono stati 30 contro 12 potenziali danni extrapiramidali da farmaci.
3.5. Altri documenti HCJE
HCJE è stato uno studio di 19 settimane, ma quando abbiamo cercato su PubMed e su clinicaltrials.gov (dove lo studio non era registrato) utilizzando il nome di Emslie, abbiamo trovato solo pubblicazioni spin-off dei dati di 9 settimane, in cui HCJE è descritto come uno studio di 9 settimane [15,16], come anche nel foglietto illustrativo approvato dalla FDA [17].
Due dei documenti spin-off affermavano erroneamente che 309 pazienti erano stati randomizzati [18,19]. Un terzo documento ha menzionato 315 pazienti e che un paziente in X065 e cinque in HCJE sono stati esclusi perché non avevano una visita post-randomizzazione [15]. Tuttavia, solo quattro pazienti in HCJE non hanno avuto una visita post-randomizzazione [10].
Dopo ulteriori ricerche, abbiamo scoperto che alcuni dei risultati di 19 settimane erano stati pubblicati, ma senza Emslie come autore [14,20]. Come notato sopra, i risultati di efficacia a 19 settimane erano meno positivi per la fluoxetina rispetto ai risultati a 9 settimane, ma sembrano non essere mai stati pubblicati per intero. Sono stati pubblicati solo i risultati per 29 non responder randomizzati nuovamente dopo 9 settimane per continuare con 20 mg di fluoxetina o per aumentare la dose fino a un massimo di 60 mg [20]. Questo è il 13% di quelli originariamente randomizzati. Gli autori hanno menzionato solo quattro dei numerosi risultati di efficacia e hanno concluso che “un aumento della dose può giovare ad alcuni pazienti” anche se non ci sono state differenze significative: risposta CDRS-R (P = 0,13), punteggio CDRS-R (P = 0,099), CGI-Gravità (P = 0,40) e CGI-Miglioramento (P = 0,30).
Un paziente in trattamento con fluoxetina 60 mg/die ha riportato un comportamento automutilatorio di lieve gravità e ha interrotto lo studio poco dopo “per mancanza di efficacia [20]”. Questo evento non è stato descritto nel rapporto di studio di 2549 pagine. Gli autori hanno concluso che gli eventi avversi erano simili nei due gruppi, ma li conteggiavano solo se erano peggiori rispetto al precedente periodo di 9 settimane. Nella discussione, sono stati più cauti e hanno notato che l’aumento del dosaggio “non era associato a un marcato aumento del numero o della gravità degli eventi avversi”. Tuttavia, non hanno spiegato cosa intendessero per “aumento marcato” o descritto la gravità degli eventi avversi.
L’ultima fase di HCJE, lo studio di prevenzione delle ricadute di 32 settimane, è stata pubblicata da Emslie et al. nel 2004 [21]. Questa volta, Emslie ha chiamato HCJE uno studio di 51 settimane. Il tempo medio alla ricaduta era più lungo in 20 pazienti che continuavano la fluoxetina rispetto a 20 pazienti passati bruscamente al placebo (P = 0,046) [21]. L’azienda sapeva già da uno studio precedente che la sospensione improvvisa degli antidepressivi poteva causare depressioni dell’astinenza in molti pazienti [22].
Una meta-analisi Lilly del 2007 sugli eventi violenti includeva tutti gli studi controllati con placebo sulla fluoxetina intrapresi in bambini e adolescenti (376 pazienti con fluoxetina e 255 con placebo) [23]. Potenziali eventi legati all’aggressione o all’ostilità sono stati identificati da una ricerca computerizzata di stringhe di testo di tutti gli eventi avversi registrati dallo sperimentatore, tutti gli eventi avversi codificati e le narrazioni. Le stringhe di testo erano composte da 71 parole o abbreviazioni e gli eventi sono stati esaminati e classificati alla cieca dal personale dell’azienda. Dato questo sforzo globale, è del tutto inverosimile che gli eventi legati all’aggressività o all’ostilità siano stati vissuti da un minor numero di bambini e adolescenti trattati con fluoxetina, 2,1%, rispetto a quelli trattati con placebo, 3,1% (P = 0,59).
Questi risultati contraddicevano i nostri risultati e la valutazione della FDA sulla domanda di Lilly per il trattamento di bambini e adolescenti con fluoxetina. La FDA ha creato una tabella delle interruzioni a causa di eventi avversi in X065, HCJE e HCJW, uno studio sul disturbo ossessivo-compulsivo che ha confrontato fluoxetina 10-60 mg al giorno con placebo per 13 settimane in 71 rispetto a 32 pazienti [13]. Ci sono state 14 contro 3 interruzioni (P = 0,02, il nostro calcolo) tra i 228 contro 190 pazienti per motivi legati al suicidio e alla violenza (tentativo di suicidio, euforia, reazione maniacale, agitazione, ipercinesia, nervosismo, disturbo di personalità, ostilità e depressione). In questi studi, ci sono stati 3 tentativi di suicidio con fluoxetina e 1 con placebo, e un altro paziente con fluoxetina è stato ricoverato in ospedale per suicidio. Sei pazienti (2,6%) in trattamento con fluoxetina hanno sviluppato mania o ipomania rispetto a nessuno con placebo (P = 0,03) [13]. Il revisore della FDA ha osservato che la mania e l’ipomania sembravano essere più comuni sulla fluoxetina in questi studi rispetto agli studi clinici sugli adulti. Una tabella degli eventi avversi segnalati spontaneamente in HCJW e HCJE (dati a 9 settimane) hanno mostrato che più pazienti hanno sviluppato ipercinesia con fluoxetina rispetto al placebo, 12 contro 1 paziente (P = 0,008, il nostro calcolo) [13].
4. Discussione
Sia Eli Lilly che Emslie et al. conclusero per entrambi gli studi che la fluoxetina è sicura ed efficace per bambini e adolescenti depressi [7-10]. Come già notato, in assenza di dati a livello di singolo paziente registrati dagli sperimentatori, non è possibile stimare l’intera scala degli effetti della fluoxetina. Tuttavia, anche in assenza di dati completi, abbiamo riscontrato che la fluoxetina non è sicura e inefficace.
4.1. Commenti sui risultati
Le valutazioni dei pazienti non hanno trovato la fluoxetina efficace e gli effetti riportati da Lilly non erano clinicamente rilevanti. L’effetto sul CDSR-R rispetto ai valori basali (58,2 e 56,2 rispettivamente in X065 e HJCE) è stato del 4% in entrambi gli studi (16% contro 9% se si utilizza LOCF). In confronto, l’effetto meno riconoscibile sulla scala equivalente per adulti, la scala della depressione di Hamilton è risultato essere 5–6 [24], corrispondente al 28% di una linea di base mediana di 25,4 in 35 studi controllati con placebo [25]. Inoltre, c’erano solo piccole differenze nel numero di pazienti che avevano sintomi minimi, o si erano ripresi o avevano un buon esito sull’indice di efficacia CGI, che confronta i benefici e i danni per ciascun paziente.
La società ha violato i suoi protocolli di prova; le informazioni essenziali erano sparse nei rapporti; mancava molto nonostante fosse indicizzato; c’erano molti errori, inspiegabili incongruenze numeriche ed esclusioni inspiegabili di pazienti dalle analisi; e i risultati che non erano coerenti con la conclusione che la fluoxetina è sicura ed efficace sono stati messi da parte o spiegati in modo inquietante.
I test statistici sono stati eseguiti all’estremo. Abbiamo trovato 5.910 test di significatività con valori P nelle CSR. Per i risultati di efficacia, il 39% era significativamente a favore della fluoxetina, nascondendo una mancata ricerca di significato sugli endpoint primari [1]. Se la fluoxetina fosse stata innocua come il placebo, 229 (5%) dei 4.575 test per gli eventi avversi sarebbero stati statisticamente significativi per caso. Con un farmaco attivo, più risultati di 229 avrebbero dovuto essere significativi, ma erano solo 174 (4%). Molti test sono stati eseguiti su eventi che si sono verificati solo in uno o due pazienti.
Nel 2002 il revisore della FDA della domanda di licenza per la fluoxetina ha notato la mancanza di efficacia negli endpoint primari e i rischi significativi per la sicurezza [1]. La FDA ha criticato Lilly per non aver cercato nel loro database segnali di eventi avversi insoliti in bambini e adolescenti. In risposta, Lilly ha fornito una revisione della letteratura per la FDA, che riguardava solo l’efficacia [13].
Lilly non ha accettato che fosse necessario un ripristino dei due studi, sostenendo nella loro lettera che avevano “analizzato adeguatamente i dati” e che non avevamo segnalato un “rischio significativamente aumentato di suicidio o comportamento aggressivo” in questi studi in la nostra meta-analisi del 2016 delle CSR degli antidepressivi [26]. Tuttavia, l’aumento del rischio di suicidio e violenza è un effetto di classe difficile da rilevare in due piccoli processi, e lo abbiamo documentato. Gli odds ratio per bambini e adolescenti erano 2,39 (intervallo di confidenza al 95% da 1,31 a 4,33) per il suicidio e 2,79 (da 1,62 a 4,81) per l’aggressività. I resoconti sommari del processo sul sito web di Lilly per fluoxetina e duloxetina sono seriamente fuorvianti, poiché il 90% dei tentativi di suicidio, tutti gli eventi di ideazione suicidaria e la maggior parte dei casi di aggressione e acatisia sono mancanti [26].
Lilly ha affermato che “la depressione è una malattia organica che risponde prontamente al trattamento” e che “l’introduzione di trattamenti antidepressivi efficaci in una fase precoce della progressione dello stato patologico ha il potenziale per trattare e controllare efficacemente la malattia, nonché per migliorare il funzionamento quotidiano e qualità della vita” [10]. Non ci sono prove che uno di questi due sia vero [5,27]. Per quanto riguarda la qualità della vita, esiste un grado estremo di segnalazioni selettive non solo nella letteratura pubblicata [28] ma anche all’interno delle CSR degli studi controllati con placebo [29]. Questi farmaci probabilmente riducono la qualità della vita, ad es. Il 12% in più di pazienti abbandona i farmaci rispetto al placebo [30] e interrompono la vita sessuale in circa la metà di quelli trattati [31], che possono continuare molto tempo dopo l’interruzione del trattamento [32].
Questo è un problema significativo per i giovani che attraversano la pubertà. Il foglietto illustrativo della fluoxetina menziona che una significativa tossicità sul tessuto muscolare, sul neurocomportamento, sugli organi riproduttivi e sullo sviluppo osseo è stata osservata in ratti giovani, con degenerazione e necrosi testicolare, vacuolizzazione dell’epididimo e ipospermia [17]. I risultati indicano che gli effetti del farmaco sugli organi riproduttivi sono irreversibili e quando gli animali sono stati valutati dopo un periodo di inattività (fino a 11 settimane dopo l’interruzione della somministrazione), la fluoxetina è stata associata ad anomalie neurocomportamentali con ridotta reattività a dosi corrispondenti a solo 10 –20% della dose umana massima raccomandata.
Il foglietto illustrativo rileva inoltre che “non esistono studi che valutino direttamente gli effetti a lungo termine della fluoxetina sulla crescita, lo sviluppo e la maturazione di bambini e adolescenti pazienti” [17]. Se estrapolato dai dati di prova, il danno corrisponde ad una perdita annuale di altezza e ad un aumento di peso rispettivamente 2,7 cm e 3,0 kg. Non sappiamo se la fluoxetina abbia anche effetti deleteri sul cervello in via di sviluppo. La FDA ha chiesto a Lilly di condurre uno studio di un anno sull’effetto della fluoxetina sulla crescita, cosa che l’azienda ha rifiutato di fare [13].
C’è stato un aumento significativo dell’intervallo QT. Lilly ha affermato che solo un paziente aveva un intervallo QT che aumentava di oltre 60 msec e che “questo paziente non ha avuto eventi avversi di interesse clinico” [14]. La FDA ha chiesto a Lilly di cercare nel database degli eventi avversi, che, nel 2001, nel gruppo di 6-17 anni, ha prodotto 7 segnalazioni di intervallo QT prolungato, 3 segnalazioni di arresto cardiaco, 1 morte improvvisa inspiegabile e 1 fibrillazione ventricolare [13].
Anche il consenso informato era problematico. Molti danni sono stati elencati nel modulo di consenso dei genitori per il processo HJCE, ma non c’era nulla sull’aumento del rischio di suicidio e violenza. Inoltre, anche se alcuni pazienti sono passati bruscamente al placebo, non c’era alcun avviso sui sintomi di astinenza, né sul fatto che l’astinenza potesse portare al suicidio e alla violenza. A proposito di rischi, è stato spiegato che la pelle e gli occhi potrebbero ingiallire, ma non che questo sia un segno di danno epatico.
Per la prova X065, sono state utilizzate 34 parole per descrivere che una puntura con un ago potrebbe causare un piccolo livido che dovrebbe causare poco o nessun disagio, mentre solo 20 parole sono state utilizzate per menzionare effetti avversi concreti della fluoxetina, senza alcun accenno che potessero essere gravi .
Gli antidepressivi raddoppiano i tentativi di suicidio sia nei bambini che negli adulti [4,5,27,33] e quindi probabilmente anche i suicidi. Il foglietto illustrativo del 2016 per la fluoxetina sottolinea quanto siano pericolosi questi farmaci [17]. Una meta-analisi di 24 studi controllati con placebo su oltre 4400 bambini e adolescenti ha mostrato che per ogni 1000 pazienti trattati con il farmaco invece del placebo, si sono verificati 14 casi aggiuntivi di suicidio (con la più alta incidenza nella depressione). Il numero necessario per nuocere è quindi solo 71.
Molti importanti professori di psichiatria e portavoce dei medici generici che sostengono l’uso di antidepressivi affermano di proteggere i bambini e gli adolescenti dal suicidio [5,34]. Anche i siti web sono fuorvianti. Un’analisi del 2018 ha mostrato che 25 (64%) su 39 siti Web popolari di 10 paesi hanno affermato che gli antidepressivi possono causare idee suicide, ma 23 (92%) contenevano informazioni errate e talvolta addirittura pericolose [35].
Ci sono quattro ragioni principali per cui la disinformazione continua. In primo luogo, molti eventi suicidi sono stati omessi o nascosti nei rapporti di prova pubblicati [4–6,26,36]. In secondo luogo, alcuni ricercatori non li hanno cercati [36]. Nonostante questi ostacoli, una meta-analisi del 2005 di studi pubblicati che includeva tutte le età ha riportato il doppio dei tentativi di suicidio con il farmaco rispetto al placebo (odds ratio 2,28, 1,14 a 4,55) [36]. Terzo, gli eventi che si verificano poco dopo l’interruzione del trattamento attivo non sono stati inclusi fino a poco tempo fa [33]. In quarto luogo, vi è un interesse commerciale a sostenere la disinformazione.
Per fortuna si può fare qualcosa. L’uso di antidepressivi nei bambini e negli adolescenti è aumentato del 59% in Danimarca dal 2006 al 2010, ma nei sei anni successivi, quando uno di noi ha costantemente informato i medici e il pubblico in generale in Danimarca del rischio di suicidio degli antidepressivi, in interviste e articoli, l’utilizzo è diminuito del 41% mentre è aumentato del 40% in Norvegia e dell’82% in Svezia nello stesso periodo [34].
Quando X065 e HCJE sono stati gestiti, c’era l’aspettativa che molti bambini infelici potessero essere aiutati con la psicoterapia e altri tipi di supporto. Questo è ancora il caso e una meta-analisi ha mostrato che la terapia cognitivo comportamentale ha dimezzato il rischio di futuri tentativi di suicidio nei giovani ammessi dopo un tentativo di suicidio [37].
4.2. Commenti sul contesto
Dato il riconoscimento da parte della FDA della mancanza di efficacia sugli endpoint primari [1] e dei significativi rischi per la sicurezza della fluoxetina, i risultati del nostro articolo sollevano tante domande sull’azienda e sulla situazione normativa intorno al 2002, così come sull’uso della fluoxetina per i minori. Il contesto in cui si sono svolti questi processi può gettare luce su questi temi.
Prima dello sviluppo degli SSRI, c’erano stati 15 studi randomizzati di antidepressivi triciclici e correlati in bambini e adolescenti, tutti negativi [38]. Negativo anche un primo studio sulla fluoxetina [39]. Nessuno di questi erano studi di alta qualità. C’era speranza che un processo ben fatto potesse dimostrare un beneficio.
C’era anche, tuttavia, un consenso clinico e letteratura sul fatto che i bambini non soffrissero di depressione endogena. Potrebbero essere infelici e infelici, ma questo era situazionale e avrebbe risposto agli interventi di supporto. Collegato a questo, non c’erano quasi psichiatri infantili con esperienza in psicofarmacologia.
Un’ulteriore caratteristica degli SSRI a quel tempo è che non erano efficaci in nessun gruppo di età per la depressione endogena (melancolia). Avevano un’azione ansiolitica, o serena. Gli SSRI sono diventati antidepressivi in parte per aggirare le preoccupazioni cliniche che qualsiasi nuovo ansiolitico avrebbe necessariamente prodotto dipendenza come avevano fatto le benzodiazepine [40]. Gli studi randomizzati condotti sui bambini supportano il punto che i farmaci sono essenzialmente ansiolitici piuttosto che antidepressivi. Una meta-analisi di studi pubblicati ha mostrato effetti significativamente maggiori per l’ansia (g = 0,56) e il disturbo ossessivo-compulsivo (g = 0,39) rispetto alla depressione (g = 0,20) [41].
Nel 2008, Erick Turner e colleghi hanno notato che il 31% degli studi sugli adulti condotti come parte di una domanda di licenza per SSRI e relativi antidepressivi considerati negativi o discutibili dalla FDA sono stati pubblicati come positivi e la dimensione dell’effetto negli articoli pubblicati era del 32% superiore rispetto alle revisioni della FDA [42]. La FDA non ha rilasciato commenti su questi risultati.
Nel 1997, anno in cui X065 è stato completato, il Congresso ha offerto sei mesi di estensione del brevetto alle aziende che hanno presentato alla FDA prove fatte sui bambini. Gli studi non dovevano essere positivi o essere pubblicati; l’intenzione dichiarata era quella di aiutare a stabilire il profilo di sicurezza nei bambini [43].
Sono necessarie due prove positive per una licenza e Lilly iniziò immediatamente a studiare HCJE. Entrambi gli studi sono stati presentati alla FDA a sostegno di un’estensione del brevetto. La FDA lo ha sostenuto e ha anche autorizzato le affermazioni secondo cui la fluoxetina potrebbe essere usata per curare i bambini depressi, come fecero altre autorità di regolamentazione quell’anno.
Dopo aver autorizzato l’affermazione della fluoxetina, basata su studi negativi sull’endpoint primario, la FDA ha emesso una lettera di approvazione nell’ottobre 2002 per la paroxetina nel trattamento di bambini e adolescenti che erano depresso. La lettera concordava con GlaxoSmithKline (GSK) che tutte e tre le prove presentate (protocolli 329, 377 e 701) nella domanda erano negative per quanto riguarda l’efficacia (lettera disponibile su study329.org). La FDA ha anche osservato: “Dato che si vedono spesso sperimentazioni negative, anche per farmaci antidepressivi che sappiamo essere efficaci, siamo d’accordo sul fatto che non sarebbe utile descrivere queste prove negative nell’etichettatura”.
Nella pubblicazione iniziale del 2001 dello studio 329, uno studio sulla paroxetina in minori depressi, GSK ha affermato che la paroxetina era sicura ed efficace [44]. Lo studio non era dissimile dagli studi sulla fluoxetina in termini di sicurezza ed efficacia. Un documento interno del 1998, tuttavia, chiariva che GSK sapeva che lo studio aveva dimostrato che il suo farmaco era inefficace, ma che sarebbe stato commercialmente inaccettabile pubblicarlo [45,46]. Il documento afferma che “i pezzi positivi dello studio sarebbero stati pubblicati” (disponibile su study329.org).
Sulla base di queste informazioni, il procuratore generale dello Stato di New York ha intentato un’azione per frode contro GSK. La definizione di tale azione ha consentito di accedere ai dati dello studio 329 e di ripristinarlo in un modo che ha dimostrato l’inefficacia della paroxetina e un raddoppio degli atti di eventi suicidi rispetto alla pubblicazione originale [6].
In risposta alle preoccupazioni sul rischio di suicidio degli antidepressivi in questa fascia di età, la FDA ha convocato una riunione del comitato consultivo sui farmaci psicofarmacologici nel febbraio 2004. La FDA ha affermato che nessuno dei farmaci (bar fluoxetina) ha dimostrato efficacia. La FDA ha rinviato un forte segnale di sicurezza, un raddoppio del rischio di eventi suicidi (P = 0,00005) [43], per un intervento in un’udienza a settembre [43]. Il caso di New York contro GSK presentato nel marzo 2004 ha innescato una crisi e, nella riunione di settembre, la FDA ha accettato la necessità di un avviso di scatola nera, in parte a causa dell’accettata mancanza di efficacia per gli antidepressivi in questa fascia di età. La licenza della paroxetina e di altri farmaci è stata interrotta, ma l’approvazione della fluoxetina non è stata revocata. I regolatori, allora e da allora, a parte quello australiano, che non approvava la fluoxetina nei minori, continuarono a sostenere che la fluoxetina è efficace in questa fascia di età.
C’è stato uno studio indipendente sulla fluoxetina, il National Institutes of Health’s Treatment of Adolescent Depression Study (TADS). Rivendicava l’efficacia e la sicurezza della fluoxetina, ma l’effetto non era clinicamente rilevante e si verificava il doppio degli eventi suicidi nei pazienti randomizzati a fluoxetina rispetto ai pazienti randomizzati a placebo [47,48]. In nessuno dei 15 articoli su questo studio tra il 2004 e il 2010, pubblicati dopo il Black Box Warnings su questi farmaci, sono stati affrontati questi dati sulla suicidalità della fluoxetina. La Duke University, dove sono stati depositati i dati dello studio, ha rifiutato di consegnare i moduli di eventi avversi gravi dello studio che potrebbero consentire il ripristino di questo studio [49].
Anche il set di dati di X065 e HCJE sugli intervalli QT ha un contesto normativo. Quando questi studi sono stati esaminati dalla FDA, Lilly ha presentato una domanda di licenza per la R-fluoxetina, che alla fine è stata ritirata in parte a causa di problemi di intervallo QTc [4]. Tali problemi sono chiaramente un problema con la fluoxetina, come lo sono con tutti gli SSRI. In risposta alle preoccupazioni della FDA sullo studio HCJE, Lilly ha sostenuto che l’aumento statisticamente significativo del QTc medio riscontrato con l’analisi iniziale era il prodotto della variabilità casuale [13]. Il revisore della FDA ha risposto che, con un valore P di 0,009, era improbabile che il risultato fosse prodotto dalla variabilità casuale.
Un’ulteriore questione da considerare deriva dall’ articolo di Turner et al.[42]. Se la FDA avesse dichiarato che lo studio 329 era negativo, avrebbe potuto aprire GSK a un’azione di frode e a un’ingente multa di transazione, come successivamente è risultato. È probabile che GSK e tutte le aziende ne fossero consapevoli durante le discussioni con la FDA. Non è compito delle autorità di regolamentazione dei farmaci controllare la letteratura medica ma, se la letteratura medica limita la libertà di movimento di un’autorità di regolamentazione, abbiamo uno scomodo nesso di problemi.
L’approvazione della fluoxetina per la depressione nei bambini e negli adolescenti e la pubblicazione di molti articoli poiché, spesso scritti da fantasmi, la pretesa di efficacia per un certo numero di SSRI ha spazzato via l’idea di affidarsi alla psicoterapia e ad altre forme di supporto. L’uso di antidepressivi negli adolescenti, in particolare femmine, è in forte aumento nella maggior parte dei paesi e queste droghe sembrano essere tra le droghe più comunemente usate dalle donne nella tarda adolescenza. Le cifre per l’utilizzo aumentano in parte perché la metà dei pazienti trova difficile ritirarsi da esse [27]. Questo è un problema particolare nelle donne in età fertile, data l’evidenza che questi farmaci aumentano i tassi di difetti alla nascita, anomalie comportamentali nei bambini nati da madri su di loro, nonché aborti spontanei e terminazioni volontarie [50,51].
La disponibilità della FDA a concedere in licenza le affermazioni secondo cui la fluoxetina era un antidepressivo nelle popolazioni pediatriche è stato un passo chiave nell’evoluzione di questo problema di salute pubblica, insieme al grande divario tra ciò che la letteratura accademica su questi farmaci diceva prima del 2004 e ciò che dicono i dati quando vi si accede. Dobbiamo capire come è nata questa situazione se vogliamo prevenire situazioni difficili in futuro.
Approvazione etica: Non richiesto.
Conflitto di interessi: Entrambi gli autori hanno compilato il modulo di divulgazione uniforme ICMJE all’indirizzo http://www.icmje.org/ coi_disclosure.pdf (disponibile su richiesta presso l’autore corrispondente) e dichiarano di aver lavorato come testimoni esperti per studi legali in casi di suicidio e omicidio causato da antidepressivi.
Finanziamento: Centro di supporto RIAT, Baltimora, Maryland. Premio n. N210955-1.
Contributi dell’autore: PCG ha ottenuto il finanziamento, ha scritto il protocollo, ha estratto i dati e ha scritto il primo manoscritto. DH ha giudicato ciecamente se gli eventi avversi fossero precursori del suicidio o della violenza e ha contribuito al manoscritto.
Condivisione dei dati: I rapporti sugli studi clinici sono disponibili su https://www.scientificfreedom.dk/links/
Materiali supplementari: Le tabelle supplementari sono disponibili su https://dx.doi.org/10.3233/JRS-210034.
Bibliografia
Vedi l’articolo originale disponibile a questo link

{kind=link}