
Irving Kirsch University of Connecticut, Thomas J. Moore The George Washington University School of Public Health and Health Services Alan Scoboria and Sarah S. Nicholls University of Connecticut
Pubblicato su Prevention & Treatment 2002
Traduzione in italiano a cura del Dott. Claudio Ajmone per GiùleManidaiBambini.org
Testo originale in inglese, disponibile a questo link
Abstract
Questo articolo riporta un’analisi dei dati di efficacia presentati alla U.S. Food and Drug Administration per l’approvazione dei 6 antidepressivi più prescritti approvati tra il 1987 e il 1999. Circa l’80% della risposta ai farmaci è stata duplicata nei gruppi di controllo con placebo, e la differenza media tra farmaco e placebo è stata di circa 2 punti sulla scala Hamilton Depression Scale a 17 voci (50 punti) e 21 voci (62 punti). Il miglioramento alle dosi più alte di farmaco non era diverso dal miglioramento alle dosi più basse. La proporzione della risposta al farmaco duplicata dal placebo è stata significativamente maggiore con i dati dei casi osservati (OC) che con i dati dell’ultima osservazione riportata (LOCF). Se gli effetti del farmaco e del placebo sono additivi, gli effetti farmacologici degli antidepressivi sono clinicamente trascurabili. Se non sono additivi, sono necessari disegni sperimentali alternativi per la valutazione degli antidepressivi.
Parole chiave: efficacia del farmaco, placebo, meta-analisi, depressione
Sebbene i farmaci antidepressivi siano ampiamente considerati efficaci, una recente meta-analisi di studi clinici pubblicati indica che il 75% della risposta agli antidepressivi è duplicata dal placebo (Kirsch & Sapirstein, 1998). Questi dati sono stati contestati per una serie di motivi, tra cui la restrizione delle analisi ai pazienti che avevano completato gli studi, il numero limitato di studi clinici valutati, le caratteristiche metodologiche di quegli studi, e l’uso di procedure statistiche meta-analitiche (Klein, 1998).
Il presente articolo riporta le analisi di un set di dati a cui queste obiezioni non si applicano, cioè i dati presentati alla Food and Drug Administration (FDA) degli Stati Uniti per l’approvazione di recenti farmaci antidepressivi. Abbiamo analizzato i dati di efficacia presentati alla FDA per i sei antidepressivi più prescritti approvati tra il 1987 e il 1999 (RxList: The Internet Drug Index, 1999): fluoxetina (Prozac), paroxetina (Paxil), sertralina (Zoloft), venlafaxina (Effexor), nefazodone (Serzone), e citalopram (Celexa).
Questi rappresentano tutti gli inibitori selettivi della ricaptazione della serotonina (SSRI) approvati durante il periodo dello studio, tranne uno. Il set di dati della FDA comprende le analisi dei dati di tutti i pazienti che hanno partecipato ad almeno una visita di valutazione, anche se poi hanno abbandonato prematuramente lo studio. I risultati sono riportati da tutti gli studi di efficacia ben controllati sull’uso di questi farmaci per il trattamento della depressione. I revisori medici e statistici della FDA hanno avuto accesso ai dati grezzi e hanno valutato gli studi in modo indipendente.
I risultati dei revisori primari medici e statistici sono stati verificati da almeno un altro revisore, e l’analisi è stata anche valutata da un comitato consultivo indipendente. Ancora più importante, i dati della FDA costituiscono la base sulla quale questi farmaci sono stati approvati. L’approvazione di questi farmaci implica che questi dati particolari sono abbastanza forti e affidabili da giustificare l’approvazione. Nella misura in cui questi dati sono difettosi, i farmaci non avrebbero dovuto essere approvati.
Khan, Warner e Brown (2000) hanno recentemente riportato i risultati di un’analisi parallela del database della FDA. Simile al rapporto di Kirsch e Sapirstein, la loro analisi ha rivelato che il 76% della risposta all’antidepressivo è stato duplicato dal placebo. Per diversi aspetti, le nostre analisi dei dati della FDA differiscono e integrano quelle riportate da Khan et al.
In primo luogo, anche se le informazioni su tutte le prove di efficacia per la depressione sono incluse nel database della FDA, i punteggi medi di cambiamento non sono stati riportati alla FDA per alcune prove in cui non è stata ottenuta una differenza significativa tra farmaco e placebo. Pertanto, i dati riassuntivi riportati da Khan et al. sovrastimano le differenze farmaco/placebo. Al contrario, noi forniamo una stima delle differenze farmaco/placebo che si basa su quei farmaci per i quali sono stati riportati tutti gli studi clinici, eliminando così il bias dovuto all’esclusione degli studi meno favorevoli al farmaco.
In secondo luogo, le medie riportate da Khan et al. (2000) non sono state aggiustate per la dimensione del campione. Così, agli studi con un piccolo numero di partecipanti è stato dato lo stesso peso dei dati più affidabili degli studi più grandi. Nella nostra analisi, i punteggi medi sono stati ponderati in base alla dimensione del campione, e le statistiche riassuntive sono state calcolate tra i farmaci per i quali erano disponibili dati completi.
In terzo luogo, nei dati riportati alla FDA sono stati utilizzati due metodi di contabilizzazione dell’attrito: l’ultima osservazione riportata (LOCF) e i casi osservati (OC). Nelle analisi LOCF, quando un paziente abbandona la sperimentazione, i risultati dell’ultima visita di valutazione vengono riportati come se il paziente avesse continuato fino al completamento della sperimentazione senza ulteriori cambiamenti. Nelle analisi OC, i risultati sono riportati solo per quei pazienti che stanno ancora partecipando alla fine del periodo di tempo valutato.
Poiché i pazienti che interrompono la terapia sono considerati come fallimenti del trattamento, le analisi LOCF sono ampiamente considerate per fornire un quadro più test conservativo degli effetti del farmaco, e l’analisi di Khan et al. (2000) era limitata a quei dati. Abbiamo usato il database della FDA per testare empiricamente questa ipotesi confrontando i dati LOCF e OC per tutti gli studi in cui entrambi sono stati riportati.
Infine, in molti degli studi riportati alla FDA, varie dosi fisse del farmaco attivo sono state valutate in bracci randomizzati separatamente. Trovare una relazione dose-risposta è un metodo per stabilire la presenza di effetti reali del farmaco. Inoltre, una relazione dose-risposta suggerisce che l’effetto del farmaco può essere sottostimato negli studi che coinvolgono bassi dosaggi. Pertanto, le nostre analisi includono un confronto degli effetti del trattamento alle dosi più basse impiegate negli studi a dose fissa con quelli alle dosi più alte.
Metodo
Usando il Freedom of Information Act, abbiamo ottenuto le revisioni mediche e statistiche di ogni studio clinico controllato con placebo per la depressione riportato alla FDA per l’approvazione iniziale dei sei farmaci antidepressivi più usati approvati nel periodo dello studio. Abbiamo ricevuto informazioni su 47 studi di efficacia randomizzati controllati con placebo a breve termine condotti per i sei farmaci a sostegno di un’indicazione approvata di trattamento della depressione. La ripartizione per studi di efficacia era la seguente: fluoxetina (5), paroxetina (16), sertralina (7), venlafaxina (6), nefadozone (8) e citalopram (5). I dati sugli studi di prevenzione delle ricadute non sono stati analizzati.
Al fine di generalizzare i risultati dello studio clinico a una più ampia popolazione di pazienti, i revisori della FDA hanno cercato un tasso di completamento del 70% o migliore per questi studi tipicamente di 6 settimane. Solo 4 dei 45 studi, tuttavia, hanno raggiunto questo obiettivo. I tassi di completamento non sono stati riportati per due studi. I tassi di abbandono erano paragonabili tra le condizioni del farmaco e del placebo. Di quegli studi per i quali questi tassi sono stati riportati, il 60% dei pazienti con placebo e il 63% dei pazienti con il farmaco in studio hanno completato uno studio di 4, 5, 6 o 8 settimane. Trentatré dei 42 studi sono durati 6 settimane, 6 studi sono durati 4 settimane, 2 sono durati 5 settimane e 6 sono durati 8 settimane. I pazienti sono stati valutati su base settimanale. Per la presente meta-analisi, i dati sono stati presi dall’ultima visita prima della fine dello studio.
Anche se la FDA ha approvato i farmaci per “il trattamento della depressione” non altrimenti specificato, tutti gli studi clinici, tranne uno, sono stati condotti su pazienti descritti come moderatamente o gravemente depressi (i loro punteggi medi al basale della Hamilton Depression Scale [HAM-D] variavano da 21,0 a 29,7). Uno degli studi è stato condotto su pazienti con depressione lieve (punteggio medio HAM-D al basale = 17,21). Trentanove dei 47 studi clinici si sono concentrati su pazienti ambulatoriali, 3 hanno incluso sia pazienti ricoverati che ambulatoriali, 3 sono stati condotti con pazienti anziani (compreso uno degli studi con pazienti ricoverati e ambulatoriali), e 2 sono stati condotti tra pazienti ricoverati per depressione grave. Nessun trial è stato riportato per il trattamento di bambini o adolescenti.
Dopo 2 settimane, la sostituzione dei pazienti è stata permessa per quelli che gli investigatori hanno determinato che non stavano migliorando in tre studi con fluoxetina e nei tre studi con sertralina per i quali sono stati riportati i dati. Gli studi includevano anche un periodo di washout da 1 a 2 settimane di placebo, durante il quale ai pazienti veniva dato il placebo. Quelli i cui punteggi sono migliorati del 20% o più sono stati esclusi dallo studio. L’uso di altri farmaci psicoattivi è stato riportato in 25 studi. Nella maggior parte degli studi, un sedativo a base di cloralio idrato è stato permesso in dosi che vanno da 500 mg a 2000 mg al giorno. Altri farmaci psicoattivi erano di solito proibiti, ma è stato comunque riportato che sono stati presi in diversi studi.
Una lacuna nei dati dell’FDA è l’assenza in molte delle relazioni di deviazioni standard riportate. Questo preclude il calcolo diretto delle dimensioni dell’effetto. Calcolare le dimensioni degli effetti dividendo le differenze medie per le deviazioni standard permette ai ricercatori di combinare i risultati degli studi in cui sono state usate diverse scale di misurazione degli esiti. Tuttavia, quando la stessa scala è usata in tutti gli studi, è possibile combinare i risultati degli studi senza prima dividerli per la deviazione standard delle scale (Hunter & Schmidt, 1990). L’HAM-D era l’endpoint primario per tutti gli studi riportati in questa analisi, permettendo così il confronto diretto dei dati di risultato senza conversione in punteggi convenzionali di dimensione dell’effetto (D). L’HAM-D è una misura ampiamente utilizzata della depressione, con coefficienti di affidabilità intergiudiziale che vanno da r = .84 a r = .90 (Hamilton, 1960).
Per ogni studio clinico, abbiamo registrato il miglioramento medio dei punteggi HAM-D nei gruppi farmaco e placebo. Successivamente, il miglioramento nel gruppo placebo è stato diviso per il miglioramento nel gruppo farmaco per fornire una stima del grado di miglioramento nei pazienti trattati con il farmaco che è stato duplicato nel gruppo placebo. Poi, la media di ciascuna di queste prove, ponderata per la dimensione del campione, è stata calcolata per ogni farmaco.
Risultati
La dimensione del campione e il cambiamento medio sulla HAM-D nelle condizioni di farmaco e placebo sono presentati nella tabella 1 per ciascuno dei 38 studi clinici su cui sono stati riportati i dati LOCF.
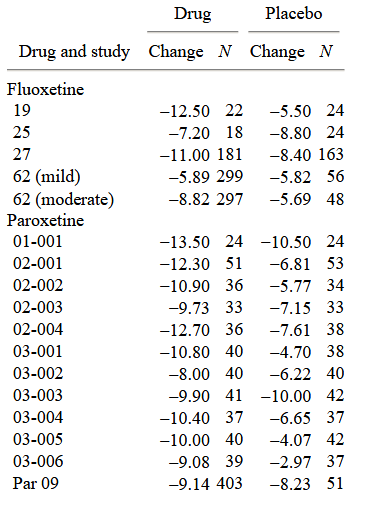
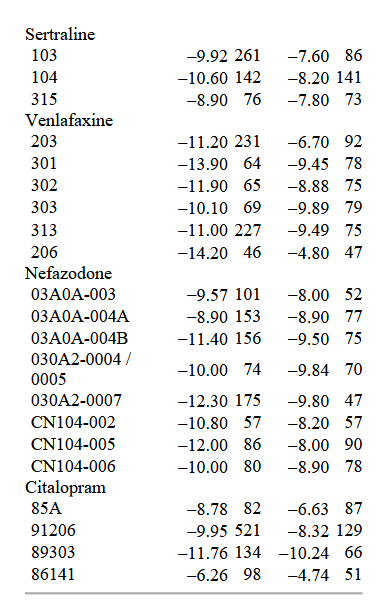
Il miglioramento medio (ponderato per la dimensione del campione) per ciascuno dei sei farmaci è presentato nella tabella 2.
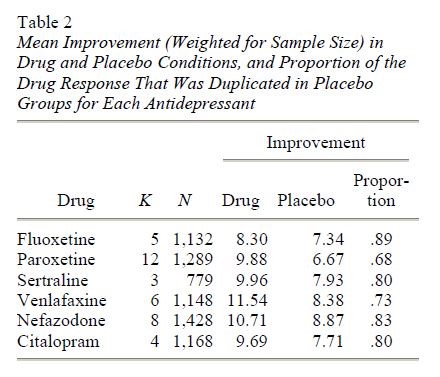
Nota. I dati non sono stati riportati da quattro studi sulla paroxetina, quattro studi sulla sertralina e uno studio sul citalopram in cui non sono state trovate differenze significative. K = numero di studi.
La versione a 17 domande dell’HAM-D è stata usata in tutti gli studi su paroxetina, sertralina, nefazodone e citalopram. La versione a 21 domande è stata usata negli studi sulla fluoxetina e sulla venlafaxina. Uno studio sul citalopram ha riportato i punteggi sia della scala a 17 voci che della scala a 21 voci, e un altro ha riportato i punteggi della scala a 17 voci e una versione a 24 voci della scala. Abbiamo usato i punteggi di 17 voci per gli studi sul citalopram perché questa versione della scala è stata usata in tutti gli studi clinici di quel farmaco. Il calcolo della risposta al farmaco e al placebo per i due studi che utilizzano forme diverse della scala rivela che il confronto farmaco/placebo è comparabile, indipendentemente dalla scala utilizzata.
I punteggi medi di miglioramento non sono stati riportati in 9 dei 47 studi. In particolare, quattro studi con paroxetina che coinvolgono 165 partecipanti, quattro studi con sertralina che coinvolgono 486 partecipanti, e uno studio con citalopram che coinvolge 274 partecipanti sono stati riportati come non riusciti a raggiungere un effetto statisticamente significativo del farmaco, ma i punteggi medi HAM-D non sono stati riportati. Questo rappresenta l’11% dei pazienti negli studi con paroxetina, il 38% dei pazienti negli studi con sertralina e il 23% dei pazienti negli studi con citalopram. In ogni caso, i revisori statistici o medici hanno dichiarato che non è stato trovato alcun effetto del farmaco.
Includere i dati degli studi con paroxetina e sertralina nelle statistiche riassuntive produrrebbe una stima gonfiata degli effetti del farmaco. Pertanto, per ottenere una stima imparziale degli effetti del farmaco e del placebo tra i farmaci, abbiamo calcolato le medie ponderate di tutti i farmaci per i quali sono stati riportati i dati di tutti gli studi clinici. Questo includeva i dati per fluoxetina, venlafaxina e nefadozone. La differenza media ponderata tra i gruppi di farmaci e placebo per questi tre farmaci è stata di 1,80 punti sull’HAM-D, e l’82% della risposta del farmaco è stata duplicata dalla risposta del placebo. Un test t, ponderato per la dimensione del campione, ha indicato che la differenza farmaco/placebo era statisticamente significativa, t(18) = 5.01, p < .001.
Nella maggior parte degli studi clinici, la dose del farmaco è stata titolata individualmente per ogni paziente entro un intervallo specificato. Tuttavia, in 12 studi che hanno coinvolto 1.942 pazienti, varie dosi fisse di un farmaco sono state valutate in bracci randomizzati separatamente. È possibile che alcune delle dosi utilizzate in questi studi fossero subcliniche. Se questo è il caso, l’inclusione di questi dati potrebbe risultare in una sottostima dell’effetto del farmaco. Per testare questa possibilità, abbiamo confrontato i dati LOCF alle dosi più basse e più alte riportate in ogni studio. In questi 12 studi, il miglioramento medio (ponderato per la dimensione del campione) era di 9,57 punti sulla HAM-D alla dose più bassa valutata e 9,97 alla dose più alta. Questa differenza tra alte e basse dosi di farmaci antidepressivi non era statisticamente significativa.
Infine, abbiamo testato l’ipotesi che le analisi LOCF forniscono test più conservativi degli effetti dei farmaci rispetto alle analisi OC. Le medie LOCF sono state riportate per tutti i 38 dei 46 studi in cui sono state riportate le medie di qualsiasi tipo. I mezzi OC sono stati riportati per 27 di questi 38 studi. In 22 studi, la differenza tra il farmaco e il gruppo placebo non era statisticamente significativa né con le misure LOCF né con quelle OC. In 12 studi, la differenza era statisticamente significativa con entrambe le misure.
In 8 studi, la differenza era significativa con LOCF ma non con OC, e 4 studi sono stati segnalati per non aver mostrato alcuna differenza tra farmaco e placebo senza specificare una regola di logoramento. Per i 27 studi per i quali sono state riportate entrambe le serie di medie, i test t correlati hanno indicato che i punteggi medi di miglioramento erano significativamente maggiori con i dati OC che con i dati LOCF sia per il farmaco, t(26) = 12,46, p < .001, che per il placebo, t(26) = 10,56, p < .001, così come la proporzione della risposta del farmaco duplicata dal placebo, t(26) = 3,36, p< .01. Nei dati LOCF, il 79% della risposta del farmaco è stato duplicato nei gruppi placebo; nei dati OC, l’85% della risposta del farmaco è stato duplicato dal placebo. Pertanto, le analisi LOCF indicano una maggiore differenza farmaco/placebo rispetto alle analisi OC.
Discussione
Negli studi clinici, si presume che l’effetto del farmaco attivo sia la differenza tra la risposta del farmaco e quella del placebo. Quindi, i dati degli studi clinici della FDA indicano che il 18% della risposta al farmaco è dovuto agli effetti farmacologici del farmaco. Questo si basa sui dati LOCF, in cui l’effetto del farmaco era significativamente più forte che nei dati OC, ed è ottenuto dopo che coloro che mostrano la maggiore risposta al placebo sono esclusi dallo studio. Nel complesso, la differenza farmaco/placebo era inferiore a 2 punti sulla HAM-D, una scala valutata dal medico altamente affidabile che è stata riportata come più sensibile delle scale valutate dal paziente alle differenze farmaco/placebo (Murray, 1989).
La gamma andava da una differenza farmaco/placebo di 3 punti per la venlafaxina a una differenza di 1 punto per la fluoxetina, entrambe sulla versione a 21 voci (64 punti) della scala. Come indicato nei memorandum della FDA (Laughren, 1998; Leber, 1998), il significato clinico di queste differenze è discutibile.
La proporzione della risposta al farmaco duplicata nei gruppi placebo è maggiore nei dati degli studi clinici della FDA che nelle precedenti meta-analisi (Khan et al., 2000; Kirsch & Sapirstein, 1998).
Le differenze possono essere dovute a due fattori: bias di pubblicazione e dati mancanti. Il bias di pubblicazione è evitato nei dati FDA dal requisito che i risultati di tutti i trial per un’indicazione siano riportati. Calcolando le statistiche di riepilogo solo per i farmaci per i quali sono state riportate le medie di tutti gli studi, si aggira il problema dei dati mancanti.
Dei due metodi ampiamente utilizzati per far fronte all’attrito negli studi clinici, le analisi LOCF sono considerate le più rigorose. Il set di dati della FDA mette in discussione questo presupposto. La proporzione dell’effetto del farmaco duplicato dal placebo era significativamente più grande nel set di dati OC che nel corrispondente set di dati LOCF. Inoltre, i gradi di libertà sono necessariamente più grandi nelle analisi LOCF, rendendo così più probabile che una differenza media sia statisticamente significativa.
Nei 47 studi clinici ottenuti dalla FDA, non ci sono stati casi in cui i dati OC hanno prodotto differenze significative che non sono state rilevate nelle analisi LOCF. Tuttavia, in 8 studi, i dati LOCF hanno prodotto differenze significative che non sono state rilevate quando sono stati analizzati i dati OC. Questi dati indicano che, rispetto alle analisi LOCF, le analisi OC forniscono test più conservativi delle differenze farmaco/placebo.
Anche se le differenze medie erano piccole, la maggior parte di esse ha favorito il farmaco attivo e, nel complesso, la differenza era statisticamente significativa. Ci sono stati solo 4 studi in cui i punteggi medi di miglioramento nella condizione del placebo erano uguali o superiori a quelli della condizione del farmaco, e in nessun caso il placebo era significativamente più efficace del farmaco attivo. Questo può indicare un piccolo ma significativo effetto del farmaco. Tuttavia, è anche possibile che questa differenza tra farmaco e placebo sia un effetto placebo potenziato dovuto alla rottura del cieco.
I dati degli studi clinici sugli antidepressivi indicano che la capacità dei pazienti e dei medici di dedurre se sono stati assegnati alla condizione di farmaco o di placebo supera i livelli del caso (Rabkin et al., 1986), possibilmente a causa della maggiore comparsa di effetti collaterali nella condizione di farmaco. Sapere che uno è stato randomizzato alla condizione di farmaco attivo è probabile che aumenti l’effetto placebo, mentre la conoscenza dell’assegnazione al gruppo placebo dovrebbe diminuire il suo effetto (Fisher & Greenberg, 1993). L’aumento degli effetti del farmaco dovuto alla rottura della cecità negli studi clinici può essere piccolo, ma la valutazione del database della FDA indica che la differenza farmaco/placebo è anche molto piccola, pari a circa 2 punti sull’HAM-D.
Anche se i nostri dati suggeriscono che l’effetto dei farmaci antidepressivi è molto piccolo e di discutibile significato clinico, questa conclusione si basa sul presupposto che gli effetti del farmaco e del placebo sono additivi. Tuttavia, è anche possibile che gli effetti del farmaco antidepressivo e del placebo non siano additivi e che il vero effetto del farmaco sia maggiore della differenza farmaco/placebo. Gli studi clinici si basano sul presupposto dell’additività (Kirsch, 2000). Cioè, il farmaco è considerato efficace solo se la risposta ad esso è significativamente maggiore della risposta al placebo, e la grandezza dell’effetto del farmaco è assunta essere la differenza tra la risposta al farmaco e al placebo.
Tuttavia, le risposte al farmaco e al placebo non sono sempre additive. L’alcol e i farmaci stimolanti, per esempio, producono almeno alcuni effetti di farmaco e placebo che non sono additivi. L’alcol placebo produce effetti che non sono osservati quando l’alcol è somministrato surrettiziamente, e l’alcol produce effetti che non sono duplicati dall’alcol placebo (Hull & Bond, 1986). Gli effetti placebo e farmacologici della caffeina sono additivi per le sensazioni di allerta ma non per le sensazioni di tensione (Kirsch & Rosadino, 1993), e risultati altrettanto misti sono stati riportati per altri stimolanti (Lyerly, Ross, Krugman, & Clyde, 1964; Ross, Krugman, Lyerly, & Clyde, 1962).
Se gli effetti del farmaco antidepressivo e gli effetti placebo antidepressivi non sono additivi, gli effetti migliorativi degli antidepressivi potrebbero essere ottenuti anche se i pazienti non sapevano che il farmaco veniva somministrato. Se questo è il caso, allora i farmaci antidepressivi hanno effetti farmacologici sostanziali che sono duplicati o mascherati dal placebo. In questo caso, gli studi clinici convenzionali sono inappropriati per testare gli effetti di questi farmaci, in quanto possono portare al rifiuto di farmaci efficaci. Al contrario, se gli effetti della droga e del placebo dei farmaci antidepressivi sono additivi, allora i dati mostrano chiaramente che questi effetti sono piccoli, nella migliore delle ipotesi, e di dubbia efficacia clinica. Infine, è concepibile che gli effetti siano parzialmente additivi, con il vero effetto del farmaco che si trova da qualche parte tra questi estremi. Il problema è che non sappiamo quale di questi modelli sia più accurato perché l’ipotesi dell’additività non è mai stata testata con la mediazione antidepressiva.
Un metodo per testare l’additività è l’uso del disegno del placebo bilanciato (Marlatt & Rohsenow, 1980). In questo disegno, il consenso informato viene prima ottenuto per uno studio in cui verrà somministrato un farmaco attivo o un placebo. A metà dei partecipanti viene detto che stanno ricevendo il farmaco attivo e l’altra metà è portata a credere che non lo stiano ricevendo. In realtà, a metà dei partecipanti viene dato un farmaco attivo e a metà no. Quindi, la metà dei partecipanti è male informata su ciò che riceverà e viene interrogata dopo la partecipazione alla sperimentazione. Come mostrato nella Figura 1, ci sono quattro celle nel disegno del placebo bilanciato.

A seconda dell’assegnazione, ai partecipanti viene (a) detto che stanno ricevendo il farmaco e in effetti lo ricevono, (b) detto che stanno ricevendo il farmaco ma in realtà ricevono il placebo, (c) detto che stanno ricevendo il placebo ma in realtà ricevono il farmaco, e (d) detto che stanno ricevendo il placebo e in effetti ricevono il placebo. Questo permette una valutazione indipendente e combinata degli effetti del farmaco e del placebo.
Questo disegno è stato usato con volontari sani e ha fornito dati interessanti sugli effetti additivi e non additivi dell’alcol (Hull & Bond, 1986) e della caffeina (Kirsch & Rosadino, 1993). Non è stato usato in studi clinici, in cui il suo uso potrebbe porre un problema etico più difficile a causa dell’inganno temporaneo che è coinvolto. Tuttavia, c’è anche un rischio etico coinvolto nel non valutare l’ipotesi di additività alla base degli studi clinici. Se questo presupposto è ingiustificato, farmaci efficaci possono essere rifiutati perché i loro effetti sono mascherati da effetti placebo. Al contrario, se l’ipotesi è giustificata, allora gli antidepressivi attuali possono essere poco più che placebo attivi. Quindi, alcuni mezzi per valutare l’ipotesi di additività è un compito cruciale.
Senza l’ipotesi di additività, i dati della FDA non permettono di determinare l’efficacia dei farmaci antidepressivi. Cioè, non è possibile determinare il grado in cui la risposta antidepressiva è un effetto del farmaco e il grado in cui è un effetto placebo. Se si fa l’ipotesi che l’effetto farmaco è la differenza tra la risposta farmaco e la risposta placebo, allora è molto piccola e di discutibile valore clinico. Di gran lunga, la maggior parte del cambiamento si osserva anche tra i pazienti trattati con placebo inerte. L’agente attivo migliora questo effetto, ma in una misura che può essere clinicamente insignificante.
Questi dati sollevano domande sui criteri utilizzati dalla FDA nell’approvazione dei farmaci antidepressivi. La FDA ha richiesto risultati positivi da almeno due studi clinici controllati, ma il numero totale di prove può variare. I risultati positivi consistono in differenze farmaco/placebo statisticamente significative. Il significato clinico di queste differenze non è considerato.
I problemi associati a questi criteri sono illustrati in un memorandum del direttore della FDA Division of Neuropharmacological Drug Products (DNDP; Leber, 1998) sull’approvazione di Celexa (citalopram) per la gestione della depressione. Due studi di efficacia controllati hanno mostrato differenze significative farmaco/placebo. Altri tre “non sono riusciti a fornire risultati che confermassero i risultati positivi” (Leber, 1998, p.6).1 Questo ha portato alla conclusione che “esiste una chiara evidenza da più di un’indagine clinica adeguata e ben controllata che il citalopram esercita un effetto antidepressivo. La dimensione di questo effetto e, cosa più importante, il valore clinico di questo effetto, non è qualcosa che può essere validamente misurato, almeno non nel tipo di esperimenti condotti. Di conseguenza, l’evidenza sostanziale nel presente caso, come in tutte le altre valutazioni dell’efficacia degli antidepressivi, parla di prova di principio [enfasi aggiunta] dell’efficacia di un prodotto” (Leber, 1998, p. 7).
Allo stesso modo, il team leader del DNDP per i prodotti psichiatrici ha commentato: “Mentre è difficile giudicare il significato clinico di questa differenza, risultati simili per altri SSRI e altri antidepressivi recentemente approvati sono stati considerati sufficienti a sostenere le approvazioni di quegli altri prodotti” (Laughren, 1998, p. 6). Laughren ha notato che “mentre le ragioni dei risultati negativi per [questi studi] sono sconosciute”, circa il 25% dei pazienti in uno degli studi falliti non soddisfaceva i criteri per la depressione maggiore, e negli altri due, “c’era una sostanziale risposta al placebo, rendendo difficile distinguere il farmaco dal placebo” (Laughren, 1998, p. 4). Sulla base di queste preoccupazioni, ha concluso: “Sento che c’erano ragioni sufficienti per speculare sugli esiti negativi e, quindi, non contare questi studi contro il citalopram” (Laughren, 1998, p. 6).
Per riassumere, i dati presentati alla FDA rivelano una piccola ma significativa differenza tra farmaco antidepressivo e placebo inerte. Questa differenza può essere un vero effetto farmacologico, o può essere un artefatto associato alla rottura del cieco da parte dei pazienti degli studi clinici e degli psichiatri che valutano la gravità delle loro condizioni. Sono necessarie ulteriori ricerche per determinare quale delle due sia il caso.
In ogni caso, la differenza è relativamente piccola (circa 2 punti sull’HAM-D), e il suo significato clinico è dubbio. La ricerca è quindi necessaria per valutare l’additività degli effetti dei farmaci antidepressivi e del placebo. Se c’è un potente effetto antidepressivo, allora è mascherato da un effetto placebo non additivo, nel qual caso l’attuale metodologia di sperimentazione clinica può essere inappropriata per valutare questi farmaci, e una metodologia alternativa deve essere sviluppata. Al contrario, se l’effetto del farmaco è così piccolo come appare quando le differenze farmaco/placebo sono stimate, allora ci può essere poca giustificazione per l’uso clinico di questi farmaci. Il problema, allora, sarebbe quello di trovare un’alternativa, dato che la risposta clinica sia al farmaco che al placebo è sostanziale. Il trattamento con placebo ha il vantaggio di suscitare meno effetti collaterali. Tuttavia, l’inganno che è inerente alla somministrazione clinica di placebo inibisce il loro uso. Quindi, lo sviluppo di metodi non ingannevoli per suscitare l’effetto placebo sarebbe di grande importanza.
Bibliografia
- Fisher, S., & Greenberg, R. P. (1993). How sound is the double-blind design for evaluating psychotropic drugs. Journal of Nervous and Mental Disease, 181, 345-350.
- Hamilton, M. A. (1960). A rating scale for depression. Journal of Neurology, Neurosurgery, and Psychiatry, 23, 56-61.
- Hull, J. G., & Bond, C. F. (1986). Social and behavioral consequences of alcohol consumption and expectancy: A meta-analysis. Psychological Bulletin, 99, 347 360.
- Hunter, J. E., & Schmidt, F. L. (1990). Methods of meta-analysis: Correcting error and bias in research findings. Newbury Park, CA: Sage.
- Khan, A., Warner, H. A., & Brown, W. A. (2000). Symptom reduction and suicide risk in patients treated with placebo in antidepressant clinical trials: An analysis of the Food and Drug Administration database. Archives of General Psychiatry 57, 311-317.
- Kirsch, I. (2000). Are drug and placebo effects in depression additive? Biological Psychiatry 47, 733-73.
- Kirsch, I., & Rosadino, M. J. (1993). Do double-blind studies with informed consent yield externally valid results? An empirical test. Psychopharmacology, 110, 437-442.
- Kirsch, I., & Sapirstein, G. (1998). Listening to Prozac but hearing placebo: A meta analysis of antidepressant medication. Prevention & Treatment, 1, Article 0002a. Available on the World Wide Web: http://www.journals.apa.org/prevention/volume1/pre0010002a.html.
- Klein, D. F. (1998). Listening to meta-analysis but hearing bias. Prevention & Treatment, 1, Article 0006c. Available on the World Wide Web: http://www.journals.apa.org/prevention/volume1/pre0010006c.html.
- Laughren, T. P. (1998, March 26). Recommendation for approvable action for Celexa (citalopram) for the treatment of depression. Memoradum: Department of Health and Human Services, Public Health Service, Food and Drug Administration, Center for Drug Evaluation and Research, Washington, DC.
- Leber, P. (1998, May 4). Approvable action on Forrest Laboratories, Inc. NDA 20-822 Celexa (citalopram HBr) for the management of depression. Memoradum: Department of Health and Human Services, Public Health Service, Food and Drug Administration, Center for Drug Evaluation and Research, Washington, DC.
- Lyerly, S. B., Ross, S., Krugman, A. D., & Clyde, D. J. (1964). Drugs and placebos: The effects of instructions upon performance and mood under amphetamine sulphate and chloral hydrate. Journal of Abnormal and Social Psychology, 68, 321 327.
- Marlatt, G. A., & Rohsenow, D. J. (1980). Cognitive processes in alcohol use: Expectancy and the balanced placebo design. In N. K. Mello (Ed.), Advances in substance abuse: Behavioral and Biological Research, (pp. 159 199). Greenwich, CT: JAI Press.
- Murray, E. J. (1989). Measurement issues in the evaluation of pharmacological therapy. In S. Fisher & R. P.Greenberg (Eds), The limits of biological treatments for psychological distress: Comparisons with psychotherapy and placebo (pp. 39-67). Hillsdale, NJ: Erlbaum.
- Rabkin, J.G., Markowitz, J. S., Stewart, J. W., McGrath, P. J., Harrison, W., Quitkin, F. J., & Klein, D. F. (1986) How blind is blind? Assessment of patient and doctor medication guesses in a placebo-controlled trial of imipramine and phenelzine. Psychiatry Research, 19,75-86.
- Ross, S., Krugman, A. D., Lyerly, S. B., & Clyde, D. J. (1962). Drugs and placebos: A model design. Psychological Reports, 10, 383 392.
- RxList: The Internet Drug Index. (1999). The top 200 prescriptions for 1999 by number of U.S. prescriptions dispensed. Retrieved November 19, 2001, from http://www.rxlist.com/99top.htm
Nota a piè di pagina
1Dati su due studi di mantenimento sono stati riportati anche dal produttore di Celexa. In questi studi di prevenzione delle ricadute, i partecipanti che avevano risposto al citalopram sono stati ramdomizzati al farmaco o al placebo. I punteggi HAM-D non distinguono tra farmaco e placebo in uno di questi studi e non sono stati valutati nell’altro. L’esito primario in questi studi era il tempo alla ricaduta (Laughren, 1998). Il tempo medio alla ricaduta è stato di 21 settimane per il citalopram contro 18 settimane per il placebo in uno di questi studi e non è stato riportato nell’altro.
